
Lesch-Nyhan综合征1例的基因突变分析
2023-02-01 12:45张小溪孙文君
安徽医药 2023年2期
张小溪,孙文君
Lesch-Nyhan综合征(Lesch-Nyhan syndrome,LNS)是由于次黄嘌呤磷酸核糖转移酶(HPRT)的突变,导致次黄嘌呤和鸟嘌呤不能转换为次黄嘌呤核苷酸(IMP)和鸟嘌呤核苷酸(GMP),而是降解为尿酸,临床多出现无法纠正的高尿酸血症,是一种罕见的X连锁隐性遗传性疾病,位于Xq26染色体上。男性多发。在美国,其发生率约为 1/380 000[1],目前,该病相关的遗传学特征及基因突变位点认识仍有限,现回顾性分析2021年7月武汉科技大学附属天佑医院收治的1例以尿酸异常升高为主要表现的LNS病儿临床资料,通过研究该病儿临床表型及遗传学特点,提高对LNS的认识。本研究符合《世界医学协会赫尔辛基宣言》相关要求。
1 临床资料
男,6月龄,因“6月竖颈不稳”就诊,病儿自3月龄起发现竖颈不稳,拉起时头颈后仰,容易伸吐舌头,情绪变化时易诱发,诉无故哭吵较为频繁,毛发生长缓慢。病儿系第3胎第3产,足月顺产。出生体质量3.2 kg,第一胎和第二胎均为顺产女婴,病儿双亲体健,为非近亲关系。否认特殊家族遗传病史。体格检查:身长60 cm,体质量6.5 kg,头围40 cm,神清,左侧眼睑下垂,竖颈不稳,双手握拳,拇指内收,四肢肌张力增高,双侧膝腱反射阳性。
实验室检查:血常规、血气分析未见明显异常,肝功能及电解质未见明显异常,肾功能:尿酸620 μmol/L,双侧大脑半球体感诱发电位(SEP)异常,左耳侧脑干听觉诱发电位(BAEP)异常,事件相关电位(ERP)异常,液相串联质谱法检测血氨基酸和酰基肉碱及尿有机酸均未见明显异常。颅脑磁共振成像(MRI)提示双侧脑室稍增宽。病儿间断在武汉科技大学附属天佑医院神经康复科行康复治疗,康复治疗期间多次监测肾功能发现血尿酸明显升高,予调整母亲饮食,碱化尿液等治疗后复查肾功能提示尿酸水平无明显下降。
结合病儿临床表现及实验室检查结果,考虑病儿存在遗传代谢相关性疾病可能,经病儿近亲属同意并签署知情同意书,采集病儿及其父母外周血送基因检测,应用高通量测序法筛查、对目标序列进行PCR后,进一步进行Sanger测序验证,并经序列分析软件得到验证结果。发现病儿HPRT1基因的外显子3(c.301(exon3)A>T)突变(编码区第301位的碱基A突变为T),为新发突变位点,氨基酸改变为P.R101X,118(该基因编码的蛋白的第101氨基酸位置上,氨基酸R突变为终止密码子,且相对原来缩短118个氨基酸长度)。病儿为半合子,父亲为野生型,母亲为杂合子(表1)。
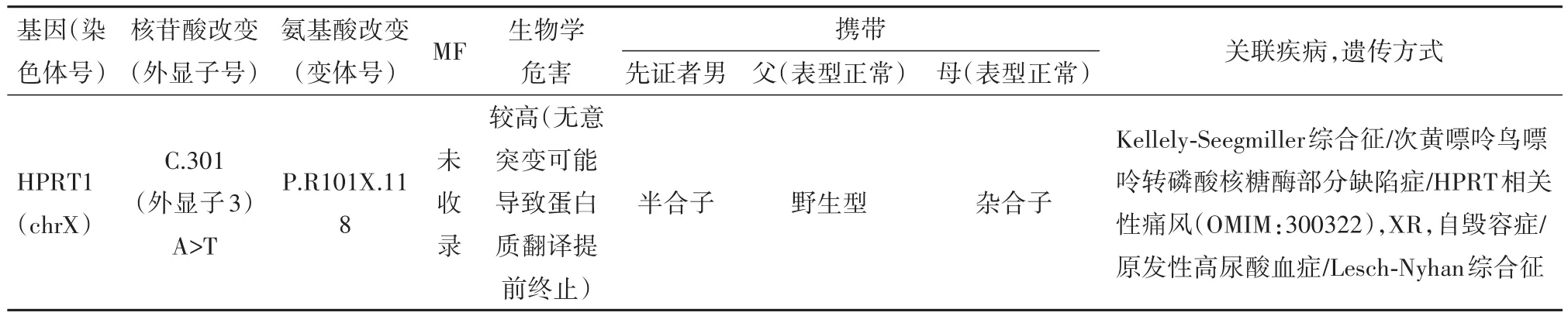
表1 病儿HRPT1基因突变及其父母表型结果
2 讨论
LNS的经典表型在1964年由Michael Lesch和William Nyhan首次报道[2],表现为HPRT功能的完全缺失[3-4],目前已知的HPRT基因突变类型有615种,包括复制、缺失、移位、插入及错义突变等。单碱基置换是最常见的基因突变类型[5]。本例报道中病儿HPRT基因突变为c.301(exon3)A>T,位于外显子3区域,是该病的一个高发突变区域,该基因编码的序列301位的碱基A突变为T,该基因编码的蛋白的第101氨基酸位置上,氨基酸R突变为终止密码子,且相对原来缩短118个氨基酸长度。上述变异会影响蛋白质功能进而致病。父母均表型正常,经Sanger测序分析验证,发现病儿父亲c.301(exo n3)位点无突变;病儿母亲c.301(exon3)A>T突变,为杂合子,该家系的基因结果符合X连锁隐性遗传规律,且该变异目前尚未见文献报道。
嘌呤核苷酸在体内的合成有2种途径,包括从头合成途径及补救合成途径。LNS即是由于补救途径中次黄嘌呤-鸟嘌呤磷酸核糖转移酶(HPRT)的缺乏,导致次黄嘌呤和鸟嘌呤不能转换为次黄嘌呤核苷酸(IMP)和鸟嘌呤核苷酸(GMP)[6],进而累积并转化为尿酸,持续的高尿酸血症会逐渐进展为痛风或肾结石,本例病儿除有顽固性高尿酸血症表现外,同时有运动发育落后,抬头不稳伴毛发生长缓慢等神经系统发育落后的症状,情绪变化时会诱发伸吐舌头及异常哭吵表现,与LNS的婴儿期临床表现相符。疾病的严重程度与突变后介导的残余酶活性密切相关[7]。除此之外,HPRT分子结构的稳定性在某些程度上同样也会影响其功能。
LNS根据临床表现可分为3型,其中经典型的临床表现包括高尿酸血症、智力迟钝、严重的运动不足、肾功能受损、急性痛风性关节炎以及反复的自残[8]。自残行为通常在2~3岁开始。也可以在生命的第一年或更晚的童年时期发展。自我伤害性行为包括反复咬嘴唇、手指和(或)手,以及对坚硬物体的反复撞击。有些孩子可能会反复挠自己的脸[9]。神经系统症状方面通常在1岁左右出现,包括手臂和腿的张力障碍,手足舞蹈徐动症等,智力迟钝也可发生,伴构音障碍和认知功能障碍[10]。LNS轻型仅表现为高尿酸血症,中间型有神经系统障碍而无自伤倾向[11]。本研究报道的这例病儿属于中间型,表现为高尿酸血症及运动发育迟缓,目前的研究发现LNS的神经损伤与HPRT酶缺乏所致基底节功能障碍及多巴胺途径有关[1]。在明显的自残行为出现前该病并无明显的特异性临床表现,使得该病的早期诊断存在一定的困难。
虽然高尿酸血症在人群中较为常见,但目前研究已发现婴儿时期高尿酸血症与遗传代谢性疾病关系密切[12],应引起重视。此外,有报道发现1例LNS病人血尿酸水平正常,但生化检查中血尿酸与肌酐比值>2[13],告诫我们在临床工作中不可忽视。
目前尚未发现有效手段治疗LNS[14]。巴洛芬或苯二氮平类药物可用于治疗痉挛等神经系统症状[15-16]。LNS病人的治疗旨在减少自残行为的行为矫正技术,如身体限制、口腔防护器等。随着年龄的增长,部分人的自残行为可能会改善或终止[17]。作为临床医生,需早期诊断并预防自残行为,进而改善病人生活质量。随着全国范围内二胎政策的开放,早期的基因检测可提前预防此类疾病的发生[18]。本例病儿目前暂未出现自残等神经系统异常的症状,主要采取碱化尿液等对症支持治疗,病儿后期的病情发展仍需进一步随访。
本例报道以持续性高尿酸血症为主要特点,提醒我们在临床诊疗过程中需警惕嘌呤代谢异常相关性疾病。基因检查及酶学分析是确诊LNS的关键[19-20]。本研究进一步丰富了对LNS病儿表型特征的认识,对于该病的诊断具有一定的参考价值,新发的突变可为遗传学研究提供更为完整的模型。
猜你喜欢
遗传(2022年6期)2022-06-21
现代食品(2022年3期)2022-03-24
安徽医药(2021年4期)2021-04-09
安徽医药(2020年1期)2020-01-10
国际呼吸杂志(2019年20期)2019-11-23
安徽医药(2019年5期)2019-05-05
精准医学杂志(2018年5期)2018-11-16
山东农业科学(2017年2期)2017-03-15
化工生产与技术(2016年5期)2016-11-07
中国运动医学杂志(2016年3期)2016-07-10
