
2-氨基-6-氯鸟嘌呤的合成工艺改进研究
2016-11-07 02:27应梦遥李坚军
化工生产与技术 2016年5期
应梦遥,李坚军
(浙江工业大学药学院,国家化学原料药合成工程技术研究中心,杭州 310014)
2-氨基-6-氯鸟嘌呤的合成工艺改进研究
应梦遥,李坚军*
(浙江工业大学药学院,国家化学原料药合成工程技术研究中心,杭州 310014)
研究了泛昔洛韦关键中间体2-氨基-6-氯鸟嘌呤的合成工艺,以鸟嘌呤为起始原料,双(三氯甲基)碳酸酯(BTC)为氯化剂进行氯化、水解反应合成目标产物2-氨基-6-氯鸟嘌呤。着重考察了反应溶剂、BTC用量和反应时间对氯化反应的影响,得到较优的工艺条件,总收率达到49%。该工艺操作简单、经济合理、环境友好,为2-氨基-6-氯鸟嘌呤的合成提供了一条新的具有潜在工业化价值的合成途径。
2-氨基-6-氯鸟嘌呤;鸟嘌呤;双(三氯甲基)碳酸酯;氯化反应
泛昔洛韦(Fanciclovir,FCV)是史克必成公司开发的第2代核苷类抗疱疹病毒药物,于1993年首先在英国上市[1]。泛昔洛韦的生物利用度高、耐受性良好,并且副作用小,在英国上市的前4个月即占据了近1/3的抗病毒药物市场[2]。近年来,随着病毒的蔓延,泛昔洛韦的需求量急剧上升。
2-氨基-6-氯鸟嘌呤是合成泛昔洛韦关键中间体,也是合成其他嘌呤类抗病毒药物(如阿巴卡韦、喷昔洛韦)的重要中间体,还可用于抗癌、降血压和消炎药物的合成[3]。其结构式为:

国内外对2-氨基-6-氯鸟嘌呤的合成工艺已有较多报道。早在1960年,Balsiger等首先报道了以鸟嘌呤为原料,与五硫化磷反应得到不稳定的硫代鸟嘌呤,再与氯气反应制得2-氨基-6-氯鸟嘌呤,该路线收率为61%,但环境污染严重,且该硫代中间体不稳定[4]。Raymond等采用鸟嘌呤与三氯氧磷为原料,在相转移催化剂(四甲基氯化铵)的作用下,直接氯化再水解制备2-氨基-6-氯鸟嘌呤,但该路线收率较低(约30%)[5]。日本Sumika公司使用鸟嘌呤与N,N-二甲基甲酰胺(DMF)在三氯氧磷的作用下,反应生成2-二甲氨基甲烯亚胺基-6-氯鸟嘌呤,再经醋酸水解、碱性水解、氨水重结晶精制得2-氨基-6-氯鸟嘌呤,总收率为55%~75%,该路线操作较为繁琐,并且使用了POCl3和氨水,对环境污染严重[6]。史克必成公司用2,5-二氨基-4,6-二氯嘧啶与氨水反应或者2,4,5-三氨基-6-羟基嘧啶在相转移催化剂存在下与磷酰氯反应,生成中间体2,4,5-三氨基-6-氯嘧啶,然后再与原甲酸三乙酯环合制备2-氨基-6-氯鸟嘌呤,收率约为62%,该方法工艺简单,但反应时间较长(>28 h)[7]。综上所述,传统的工艺路线普遍存在三废污染大、成本高等问题。
本研究在文献的基础上,设计了以鸟嘌呤为原料,双(三氯甲基)碳酸酯(BTC)为氯化剂进行氯化反应,再经过水解反应制备2-氨基-6-氯鸟嘌呤的合成路线[6]。
1 实验部分
1.1 仪器与试剂
Varian-400(400 MHz)核磁共振仪(d6-DMSO为溶剂,四甲基硅烷为内标),Büchi B-540熔点仪(温度未经校正),ZF-Ⅰ型三用紫外分析仪,Thermo Finnigan LCQ Advantage(ESI)质谱仪。
反应使用的薄层色谱(TLC)板,自制,硅胶GF254,使用前活化。鸟嘌呤为工业级,其余试剂为化学纯,未经处理直接使用。
1.2 合成实验
1.2.1 2-甲酰氨基-6-氯鸟嘌呤的合成
在250 mL三口瓶中加入DMF 4.7 mL、甲苯80mL,冰水浴中机械搅拌。缓慢滴加含有BTC(5.9 g,0.02 mol)的20 mL甲苯溶液,滴完后继续反应1 h。然后,在室温下,加入鸟嘌呤(3.0 g,0.02 mol),升温回流反应5 h。反应式为:
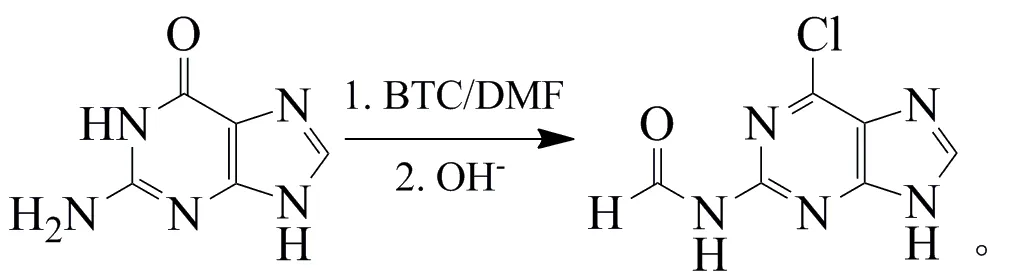
反应结束后,将上述反应液倒入100 mL冰水中,分出有机相,水相用质量分数10%的NaOH溶液调节pH至3,升温至70℃水解反应3 h,冷却至室温,抽滤,水洗,烘干,得到淡黄色固体2.74 g,收率69.3%。
熔点>260 ℃,1H NMR(400 MHz,d6-DMSO):δ 13.63(s,1H,CONH),11.95(s,1H,NH),9.32(s,1H,CHO),8.46(s,1H,CH)。MS(ESI):m/z=196.2[MH]-,198.2。
1.2.2 2-氨基-6-氯鸟嘌呤的合成
在100 mL单口瓶中加入上述中间体2-甲酰氨基-6-氯鸟嘌呤,加入50 mL质量分数10%的NaOH溶液溶解,室温下磁力搅拌2 h,反应式为:
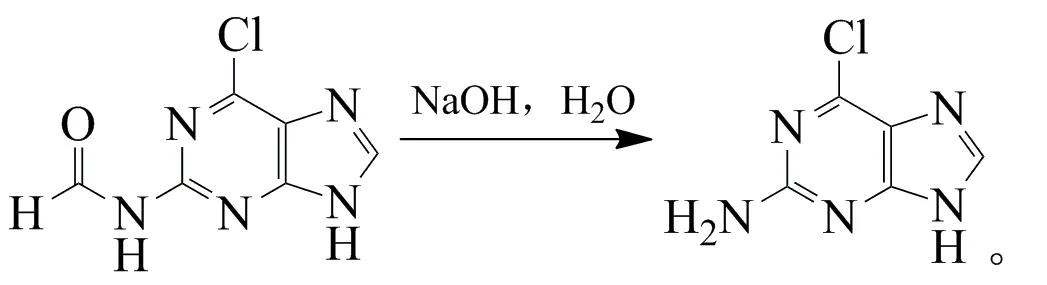
反应结束后,冷却至室温,用质量分数35%的盐酸中和,过滤,水洗得2-氨基-6-氯鸟嘌呤粗品。接着,所得粗品不用干燥,直接用质量分数5%的NaOH溶液重结晶,过滤,水洗,滤饼烘干,得白色固体1.67 g,收率71.0%。
熔点>300 ℃(文献>350 ℃[6]);1H NMR(400 MHz,d6-DMSO):δ 12.81(s,1H,NH),8.07(s,1H,CH),6.74(s,2H,NH2)。MS(ESI):m/z=168.2[MH]-,170.2。
2 结果与讨论
中间体2-甲酰胺基-6-氯鸟嘌呤的水解反应操作简单,收率相对较稳定,因此,着重对2-甲酰胺基-6-氯鸟嘌呤的合成工艺进行了讨论。
2.1 反应溶剂对反应的影响
从反应条件和溶剂效应考虑,主要选用含氯的非质子极性溶剂和沸点较高的苯类溶剂为考察对象。以鸟嘌呤为原料,与BTC、DMF反应,再经水解制备2-甲酰胺基-6-氯鸟嘌呤,考察不同的溶剂对反应的影响,结果见表1。

表1 反应溶剂对氯化反应收率的影响Tab 1 Effect of reaction solvent on chlorination reaction yield
由表1可知,选择二氯甲烷和氯仿作溶剂时几乎无目标产物生成。选用1,2-二氯乙烷为溶剂,收率较低。选用甲苯和二甲苯作溶剂,收率相对较高。综上,考虑高温对反应的影响,选择甲苯为较优的反应溶剂。
2.2 BTC用量对反应的影响
在确定了反应溶剂后,进一步对BTC用量进行了优化,结果见表2。

表2 BTC用量对氯化反应收率的影响Tab 2 Effect of BTC dosage on yield of chlorination reaction yield
由表2可知,BTC与鸟嘌呤的摩尔比为0.5时,收率较低,可能原因是氯化反应未完全进行。当BTC与鸟嘌呤的摩尔比大于1.0时,收率有了较大的提高,考虑到成本因素,选择BTC与鸟嘌呤的摩尔比1.0的BTC投料量为较优条件。
2.3 回流时间对反应的影响
按照1.2.1节中的合成方法,考察回流时间对反应收率的影响,结果见表3。

表3 回流时间对氯化反应收率的影响Tab 3 Effect of refluxing time on chlorination reaction yield
从表3可以发现,反应收率随着反应时间的增加而增加。当反应时间大于5 h后,收率并没有明显提高,因此,选择5 h为较优反应时间。
3 结论
开发了一条以鸟嘌呤为原料、使用BTC为氯化剂进行氯化反应,再经过水解反应合成2-氨基-6-氯鸟嘌呤的工艺,总收率约为49%。同时对氯化反应中的反应溶剂、BTC的用量和反应时间进行了优化,得到的较优反应条件为:以甲苯为反应溶剂,BTC与鸟嘌呤的摩尔比1.0,回流反应5 h。此工艺操作简单、安全、经济合理、环境污染小,具有潜在的工业化价值。
[1]刘自贵.抗病毒感染药物治疗进展[J].现代临床医学,2015,41(6):468-475.
[2]关维先,关月秋.嘌呤类抗病毒药物的进展[J].中国医药情报,1995,1(3):182-185.
[3]Schinazi R F,Keane T E,Liotta D C.Treatment of drogenital cancer with born netron capture therapy,and preparation of carboranyl-containingnucleosides:WO9614073[P].1996-05-17.
[4]Balsiger R W,Montgomery J A.Synthesis of potential anticancer agents.XXV.Preparation of 6-alkoxy-2-aminopurines[J].J Org Chem,1960,25:1573-1575.
[5]Raymond H M,Lewis J R.Process for the preparation of 2-amino-6-chloro-purine:EP0203685A2[P].1986-12-03.
[6]Igi M,Hayashi T.Method for producing 2-amino-6-halogenopurine and synthesis intermediate therefor:EP0543095A2[P].1993-05-26.
[7]Christopher H J.Process for preparation of 2-amino-6-chloropurine:WO9407892[P].1994-04-14.
TQ251.3+4
ADOI10.3969/j.issn.1006-6829.2016.05.009
*通讯联系人。电子邮件:lijianjun@zjut.edu.cn
2016-05-18
猜你喜欢
遗传(2022年6期)2022-06-21
中国药学药品知识仓库(2022年10期)2022-05-29
皮肤病与性病(2021年3期)2021-07-30
汕头大学学报(自然科学版)(2020年4期)2020-12-14
医药前沿(2020年12期)2020-12-04
中成药(2017年5期)2017-06-13
中国运动医学杂志(2016年3期)2016-07-10
化工设计通讯(2016年12期)2016-03-01
中国继续医学教育(2015年6期)2016-01-07
股市动态分析(2015年12期)2015-09-10
