
西洋参茎叶总皂苷通过抑制内质网应激减轻毒胡萝卜素诱导的心肌细胞凋亡*
2014-08-13 12:06王晓礽陶天琪徐菲菲刘秀华史大卓
中国病理生理杂志 2014年10期
刘 蜜, 王晓礽, 陶天琪, 徐菲菲, 刘秀华△, 史大卓
(1解放军总医院病理生理研究室,北京 100853; 2中国中医科学院西苑医院,北京 100091)
心肌细胞凋亡在缺血性心脏疾病中扮演着重要角色[1]。抑制心肌细胞凋亡可以改善心脏功能[2]。内质网应激(endoplasmic reticulum stress, ERS)相关细胞凋亡途径是一种新提出的凋亡途径,缺血[3-4]、缺氧/复氧[5]、缺血再灌注损伤[6]及压力过负荷[7]等均可以激活ERS相关凋亡途径。在哺乳动物中,ERS反应主要由3种主要的内质网跨膜蛋白介导,分别是蛋白激酶R样内质网激酶(protein kinase R-like endoplasmic reticulum kinase, PERK)、活化转录因子6(activating transcription factor 6, ATF6)和需肌醇酶1(inositol-requiring enzyme 1, IRE1)。ERS的最初目的是降低未折叠蛋白水平及恢复细胞内稳态。然而,持续或严重的ERS激活促凋亡信号。内质网内主要伴侣分子葡萄糖调节蛋白78(glucose-regulated protein 78, GRP78)与这3种感受器蛋白分离,导致其激活。其中PERK-真核起始因子2α(eukaryotic initiation factor 2α, eIF2α)-ATF4-C/EBP同源蛋白(C/EBP homologous protein, CHOP)通路是最主要的一条信号通路。我们前期研究证实CHOP介导的ERS相关凋亡参与大鼠急性心肌梗死后非梗死区心肌细胞凋亡的发生[3]。
毒胡萝卜素(thapsigargin, TG)是一种倍半萜内酯,可通过抑制肌浆网/内质网Ca2+-ATP酶扰乱内质网内钙稳态,导致内质网内Ca2+耗竭以及胞浆Ca2+浓度增加[8-10]。耗竭内质网Ca2+能激发ERS,如果这种状况持续存在,则能引起ERS相关凋亡[11]。
我们前期研究发现,西洋参茎叶总皂苷(Panaxquinquefoliumsaponin, PQS)可通过抑制ERS相关凋亡,减轻离体大鼠心肌细胞缺氧/复氧损伤[5]及大鼠缺血再灌注损伤[6],并且PQS可通过抑制CHOP介导的ERS相关凋亡减轻AMI后心室重构[3]。然而,尚无PQS可以直接抑制TG诱导ERS相关细胞凋亡的报道。本研究采用TG诱导离体乳大鼠心肌细胞凋亡,并且进一步以RNA干扰(RNA interference, RNAi)方法敲低心肌细胞内质网PERK基因,探讨PQS抑制ERS相关凋亡的信号途径。
材 料 和 方 法
1 材料
出生24 h内清洁级Sprague-Dawley(SD)新生鼠由军事医学科学院实验动物中心提供;PQS粉由吉林省集安益盛药业股份有限公司提供;DMEM培养基购自Gibco;新生牛血清 (newborn calf serum,NCS)购自杭州四季青公司;胰蛋白酶(trypsin)购自Amresco;青、链霉素混合抗生素购自Gibco;蛋白酶抑制剂和牛磺酸购自Sigma;Annexin V-FITC细胞凋亡检测试剂盒购自南京凯基生物工程公司;Cell Counting Kit-8(CCK-8)购自Dojindo;蛋白电泳分子量(7~175 kD)标志物为Bio-Rad产品;兔抗人GRP78多克隆抗体、兔抗人钙网蛋白(calreticulin,CRT)多克隆抗体购自Stressgen;兔抗人GAPDH单克隆抗体、小鼠抗人CHOP单克隆抗体、兔抗人Bax和Bcl-2多克隆抗体及兔抗人PERK、eIF2α和p-eIF2α多克隆抗体均购自Cell Signal Technology;增强化学发光试剂盒购自Millipore;兔抗人p-PERK、ATF4多克隆抗体和辣根过氧化酶标记山羊抗兔及山羊抗小鼠IgG 购自Santa Cruz。
2 乳鼠心肌细胞培养
采用本实验室[14]方法,无菌操作取出生后24 h内SD新生鼠心尖部组织,剪碎成1 mm×1 mm×1 mm大小,加入适量0.15%胰蛋白酶,37 ℃水浴下轻柔搅动、反复消化,制备心肌细胞悬液,差速贴壁。用含15%新生牛血清的DMEM培养液,调整细胞浓度为每瓶3×106个,接种于底面积为75 cm2的培养瓶,置CO2培养箱进行原代培养。
3 实验分组
取原代培养心肌细胞,置于CO2培养箱常规培养24 h,换无新生牛血清的DMEM 培养液同步化24 h后,根据预实验结果,选取保护作用明显的给药浓度160 mg/L(终浓度)进行下列实验。实验分为以下5组:(1) 正常对照(control)组:细胞置于CO2培养箱37 ℃常规培养24 h至实验结束;(2) TG组:培养液内加入1 μmol/L TG,常规培养24 h至实验结束;(3) PQS+TG组:在培养液中加入PQS,使终浓度160 mg/L,预处理24 h,之后处理同 (2) 组;(4)PERK敲低+TG组(si-PERK+TG):针对rat PERK(NC_005103.3)设计并筛选出25 bp的稳定型双链小干扰RNA(stealth siRNA, stRNA)stPERK-1,以终浓度50 nmol/L转染细胞,转染方法按照Lipofectamine 2000(Invitrogen)说明书进行,转染6 h后更换含10%新生牛血清的DMEM培养液,同时加入TG使其终浓度为1 μmol/L,常规培养24 h至实验结束;(5) 随机双链RNA转染对照+ TG组 (mock+TG):随机合成的双链stealth siRNA 对细胞进行转染6 h 后更换含10%新生牛血清的DMEM培养液,同时加入TG使其终浓度为1 μmol/L,常规培养24 h至实验结束。
4 方法
4.1CCK-8法检测细胞活力 以2×103cells/well的密度接种96孔板,每孔培养液体积100 μL,每组8个复孔,重复3次。实验处理结束后加入10 μL CCK-8,以没有接种细胞而加入等量培养液和CCK-8的孔作为对照。96孔板在培养箱培养3 h后以酶标仪在450 nm下检测A值。
4.2Annexin V/PI双标法检测细胞凋亡 以1×104/cm2的密度接种细胞于60 mm培养皿,参照annexin V-FITC 凋亡检测试剂盒说明书的方法进行检测:实验处理结束后以不含EDTA的0.25%胰酶消化并收集细胞,以0.01 mol/L PBS离心洗涤细胞2次,每次的离心条件为3 000 r/min × 30 s,洗涤后的沉淀以500 μL binding buffer悬浮均匀,分别加入5 μL annexin V和5 μL PI,充分混合均匀,室温下避光孵育10 min后,以流式细胞仪(BD FACSVerse)检测早期凋亡率和晚期凋亡率之和。
4.3Westernblotting按Liu等[15]报道方法提取心肌细胞总蛋白,BCA法蛋白定量后分装,-80 ℃保存。取上述细胞蛋白提取液上清 (含蛋白80μg) 进行SDS-PAGE(12%分离胶,将电泳分离后的蛋白质电转移至硝酸纤维素膜上,用5%BSA封闭1h后分别加入GRP78、CRT、PERK、eIF2α、p-eIF2α、Bcl-2及Bax多克隆抗体(均为1∶500)、p-PERK多克隆抗体(1∶200)、CHOP单克隆抗体(1∶500)和GAPDH单克隆抗体(1∶1 000)4 ℃过夜孵育,用1×TBS-T洗膜后,以相应的Ⅱ抗孵育1.5h。化学发光ECL显示,采用Image-ProPlus4.1软件分析蛋白条带的积分吸光度值(integratedabsorbance, IA=平均吸光度值×面积),以靶蛋白IA值/GAPDHIA值的比值反映靶蛋白水平。
5 统计学处理
数据用均数±标准差(mean±SD)表示,采用 SPSS 17.0 统计软件分析。单因素方差分析(one-way ANOVA)进行多组间比较,S-N-K法进行多组间两两比较。以P<0.05为差异有统计学意义。
结 果
1 不同浓度PQS对TG诱导心肌细胞凋亡及细胞活力的影响
流式细胞术及CCK-8法检测发现:与正常对照组比较,TG处理使心肌细胞凋亡率升高31.5%(P<0.05),使心肌细胞活力降低69.2%;与TG组比较,PQS(40 mg/L、80 mg/L及160 mg/L)预处理及牛磺酸预处理可使心肌细胞凋亡率分别降低19.0%、35.2%、51.1%和54.4%(P<0.05),使心肌细胞活力分别升高3.5%、15.4%、28.5%和32.6%(P<0.05),见图1。
2 不同浓度PQS对TG诱导心肌细胞Bcl-2及 Bax表达比值的影响
Western blotting结果发现,与正常对照组比较,TG处理使心肌细胞Bcl-2与 Bax表达比值降低57.9%(P<0.05);与TG组比较,PQS(40 mg/L、80 mg/L及160 mg/L)预处理及牛磺酸预处理可使心肌细胞Bcl-2/Bax升高9.8%、13.8%、82.7%和97.9%(P<0.05),见图2。
3 PQS及敲低PERK对TG诱导心肌细胞凋亡及细胞活力的影响
流式细胞术及CCK-8法检测发现:与TG组比较,PQS预处理可使心肌细胞凋亡率降低36.2%(P<0.05),使心肌细胞活力升高47.4%(P<0.05),敲低PERK表达可使心肌细胞凋亡率降低45.2%(P<0.05),使细胞活力升高63.7%(P<0.05);与TG组比较,mock+TG组细胞凋亡率和细胞活力没有明显改变(P>0.05)。PQS预处理与PERK基因敲低对TG处理心肌细胞的保护作用无明显差异(P>0.05),见图3。
4 PQS及敲低PERK对TG诱导心肌细胞内质网应激分子表达的影响
Western blotting结果显示,与control组比较,TG可使GRP78和CRT蛋白的表达分别升高4.9倍和4.2倍(P<0.05);使PERK和eIF2α的磷酸化分别升高96.9%和82.9%(P< 0.05);使ATF4和CHOP的蛋白表达分别升高8.7倍和3.1倍(P<0.05)。与TG组比较,PQS预处理可使GRP78、CRT蛋白表达分别降低75.5%和78.4%(P<0.05);使PERK、eIF2α磷酸化分别降低36.4%和27.6%(P<0.05);使ATF4和CHOP蛋白表达分别降低75.1%和60.6% (P<0.05)。与TG组比较,敲低PERK对GRP78和CRT蛋白表达均无显著影响(P>0.05);敲低PERK可使PERK和eIF2α蛋白磷酸化分别降低33.1%和28.3%(P<0.05);敲低PERK可使ATF4和CHOP蛋白表达分别降低68.5%和32.0%(P<0.05)。与TG组比较,mock+TG组PERK及eIF2α蛋白磷酸化、GRP78、CRT、ATF4和CHOP蛋白表达没有显著改变(P<0.05)。PQS预处理与敲低PERK在降低PERK和eIF2α蛋白磷酸化以及ATF4和CHOP蛋白表达方面结果相似(P>0.05),见图4。

Figure 1. Effects of PQS on the apoptosis (A) and viability (B) of cardiomyocytes. Mean±SD. n=3.#P<0.05 vs control; *P<0.05 vs TG.
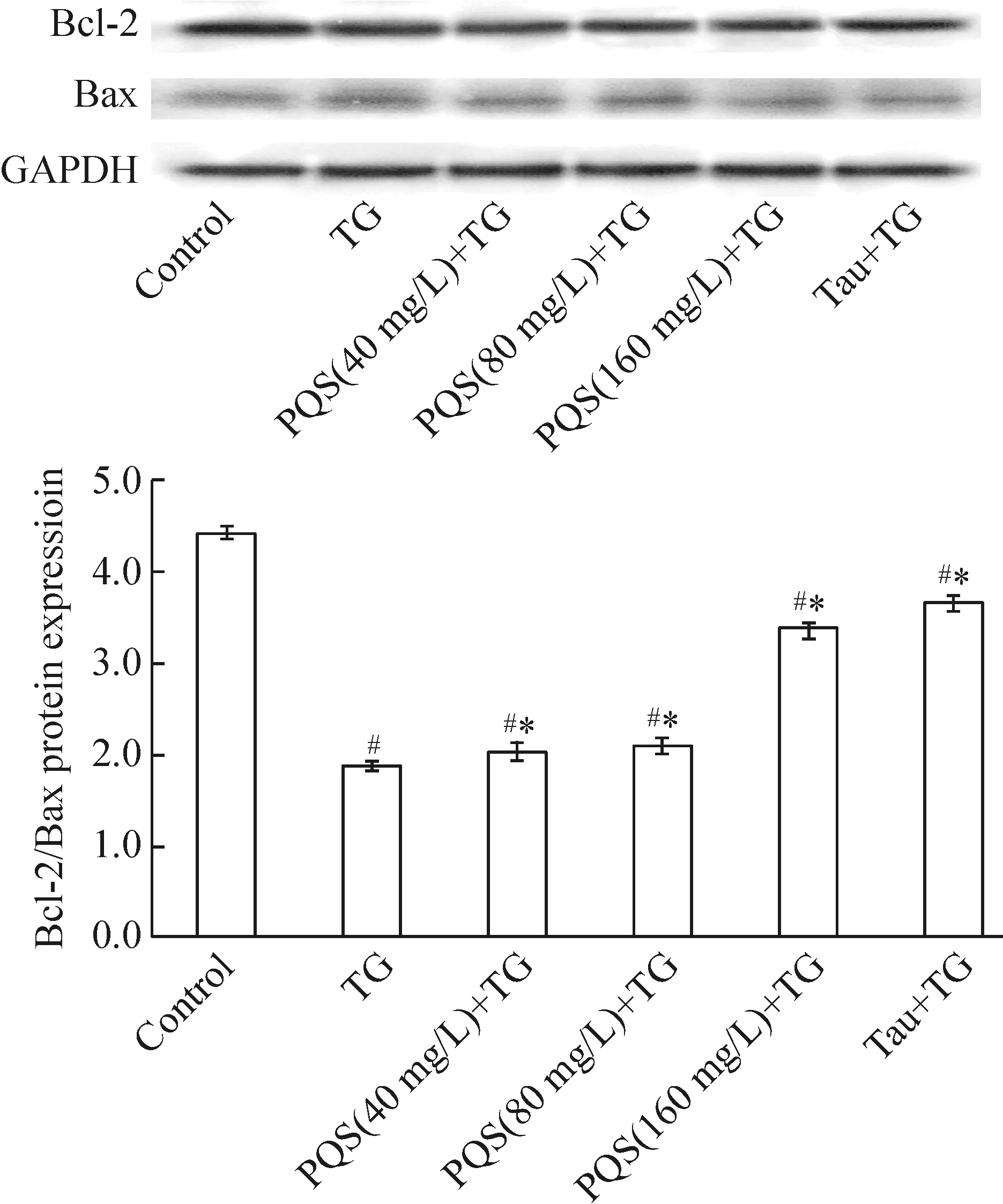
Figure 2. The effect of PQS on the expression of Bax and Bcl-2.Mean±SD.n=3.#P<0.05 vs control; *P<0.05 vs TG.
讨 论
持续ERS时PERK磷酸化,依次磷酸化eIF2α,抑制多数蛋白质合成,并激活ATF4[16]。这种ERS信号系统能诱导过表达的CHOP与其它转录因子结合,导致促凋亡基因表达[17-18]。CHOP介导的ERS相关凋亡参与了多种心血管疾病的发生发展[8, 19-21]。Zhang等[8]通过腹主动脉狭窄诱导高血压心肌肥厚的大鼠模型,腹主动脉狭窄早期即可诱导内质网分子伴侣CRT表达增加,激活ERS的适应性途径;后期可诱导ERS相关CHOP途径,导致心肌细胞凋亡。本室前期工作证实[21],TG可诱导心肌细胞的ERS反应,CRT、GRP78、ATF4和PERK表达升高:TG 1~10 nmol/L作用48 h和50 nmol/L作用12~36 h,可诱导Bcl-2/Bax比值代偿性增加,具有细胞保护作用;TG 70~100 nmol/L作用48 h或50 nmol/L作用72 h则诱导过度ERS,导致CHOP表达增加和Bcl-2/Bax比值下降,触发心肌细胞凋亡途径,表明CHOP介导的ERS相关凋亡途径参与了TG诱导的心肌细胞肥大过程。结合本室前期工作及文献报道[12-13],本研究采用1 μmol/L TG处理体外培养的乳大鼠心肌细胞24 h,与对照组比较,心肌细胞凋亡显著增加,细胞活力明显降低,且ERS分子GRP78蛋白表达、PERK磷酸化和CHOP蛋白表达显著增加,抗凋亡蛋白Bcl-2与促凋亡蛋白Bax比值明显降低,说明出现明显ERS相关凋亡,和文献[21]中TG的干预剂量和时间不同,但结果一致。

Figure 3. The effects of PQS and knockdown of PERK on the apoptosis (A) and viability (B) of cardiomyocytes. Mean±SD.n=3.#P<0.05 vs control; *P<0.05 vs TG;△P<0.05 vs mock+TG.
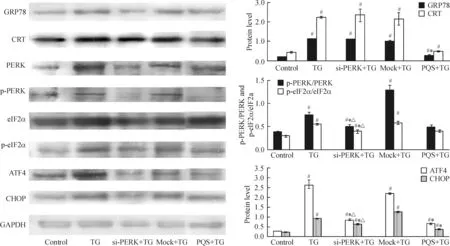
Figure 4. The effects of PQS and PERK knockdown on protein expression of ERS molecules in the cardiomycytes.Mean±SD.n=3.#P<0.05 vs control; *P<0.05 vs TG; △P<0.05 vs mock+TG.
Tsukano等[22]用0.1 μmol/L TG处理体外培养的大鼠皮质神经元24 h,证明可诱导caspase-9及caspase-3的激活,导致细胞凋亡。Szegezdi等[23]用1.5 μmol/L TG处理PC12细胞24 h,结果表明可导致细胞凋亡,机制涉及caspase-12的激活及CHOP表达增加。进一步研究发现,TG诱导PC12细胞激活Bim,诱导Bax向线粒体转位,导致线粒体释放细胞色素C,激活caspase-3、caspase-7及caspase-9,最终诱导线粒体相关凋亡。
为了证实PQS可通过抑制ERS相关凋亡发挥心血管保护作用,本研究在TG诱导之前,用PQS预处理24 h,结果表明可显著减轻TG诱导的细胞凋亡及细胞活力下降,且呈剂量依赖性地升高促凋亡蛋白Bax的表达,降低抗凋亡蛋白Bcl-2的表达。PQS 160 mg/L与ERS抑制剂牛磺酸作用相似。本课题组前期工作发现[3,5],PQS能剂量依赖性地降低心肌细胞缺氧/复氧损伤,并可通过抑制ERS相关凋亡减轻大鼠急性心肌梗死后心室重构,机制涉及PQS降低ERS分子GRP78、CRT、CHOP及Bax蛋白的表达,升高抗凋亡分子Bcl-2蛋白表达等方面。本研究结果PQS可抑制CHOP介导的ERS相关凋亡,与前期报道一致。
为进一步探讨PQS抑制ERS相关凋亡的作用途径,我们对内质网跨膜蛋白PERK表达进行了干预。通过细胞转染si-PERK敲低PERK表达,结果表明可显著降低TG诱导的心肌细胞凋亡,升高细胞活力,抑制ERS分子PERK、eIF2α、ATF4和CHOP的翻译,并可抑制PERK和eIF2α磷酸化,但对内质网伴侣分子GRP78和CRT蛋白的水平无明显影响,与文献报道一致[22, 24]。PQS预处理组与敲低PERK组在心肌细胞凋亡、心肌细胞活力及ERS分子的表达方面均无显著差异,PQS预处理可模拟敲低PERK对心肌细胞的保护作用。
综上所述,本实验证明PQS通过抑制过度ERS,发挥抗心肌细胞凋亡作用,其作用机制涉及PQS抑制PERK-eIF2α-ATF4-CHOP途径,表现为PQS可以降低PERK及eIF2α的磷酸化水平,降低TG诱导的GRP78、CRT、ATF4和CHOP蛋白表达,从而减轻ERS凋亡通路激活。
[参 考 文 献]
[1]FeuersteinGZ,YoungPR.Apoptosisincardiacdiseases:stress-andmitogen-activatedsignalingpathways[J].CardiovascRes, 2000, 45(3):560-569.
[2] Hayakawa Y, Chandra M, Miao W, et al. Inhibition of cardiac myocyte apoptosis improves cardiac function and abolishes mortality in the peripartum cardiomyopathy of Gαq transgenic mice [J]. Circulation, 2003, 108(24):3036-3041.
[3]LiuM,WangXR,WangC,etal. Panax quinquefoliumsaponinattenuatesventricularremodelingafteracutemyocardialinfarctionbyinhibitingCHOP-mediatedapoptosis[J].Shock, 2013, 40(4):339-344.
[4] Xin W, Li X, Lu X, et al. Involvement of endoplasmic reticulum stress-associated apoptosis in a heart failure model induced by chronic myocardial ischemia [J]. Int J Mol Med, 2011, 27(4): 503-509.
[5]WangC,LiYZ,WangXR,etal. Panax quinquefoliumsaponinsreducemyocardialhypoxia-reoxygenationinjurybyinhibitingexcessiveendoplasmicreticulumstress[J].Shock, 2012, 37(2):228-233.
[6] 王 琛, 刘 蜜, 孙 胜, 等. 西洋参茎叶总皂苷通过抑制过度内质网应激减轻大鼠心肌缺血/再灌注损伤[J]. 中国病理生理杂志, 2013, 29(1):20-27.
[7]LyttonJ,WestlinM,HanleyMR.ThapsigargininhibitsthesarcoplasmicorendoplasmicreticulumCa-ATPasefamilyofcalciumpumps[J].JBiolChem, 1991, 266(26):17067-17071.
[8] Zhang ZY, Liu XH, Ye YJ, et al. C/EBP homologous protein-mediated endoplasmic reticulum stress-related apoptosis pathway is involved in abdominal aortic constriction-induced myocardium hypertrophy in rats[J]. Acta Physiol Sin, 2009, 61(2):161-168.
[9]SagaraY,InesiG.InhibitionofthesarcoplasmicreticulumCa2+transportATPasebythapsigarginatsubnanomolarconcentrations[J].JBiolChem, 1991, 266(21):13503-13506.
[10] Treiman M, Caspersen C, Christensen SB. A tool coming of age: thapsigargin as an inhibitor of sarco-endoplasmic reticulum Ca2+-ATPases[J]. Trends Pharmacol Sci, 1998, 19(4):131-135.
[11]DenmeadeSR,IsaacsJT.TheSERCApumpasatherapeutictarget:makinga“smartbomb”forprostatecancer[J].CancerBiolTher, 2005, 4(1):14-22.
[12] Chung H, Chung HY, Bae CW, et al. Ghrelin suppresses tunicamycin- or thapsigargin-triggered endoplasmic reticulum stress-mediated apoptosis in primary cultured rat cortical neuronal cells[J]. Endocrine J, 2011, 58(5):409-420.
[13]FöldiI,TóthAM,SzabóZ,etal.Proteome-widestudyofendoplasmicreticulumstressinducedbythapsigargininN2aneuroblastomacells[J].NeurochemInt, 2013, 62(1): 58-69.
[14] Liu X, Xu F, Fu Y, et al. Calreticulin induces delayed cardioprotection through mitogen-activated protein kinases[J]. Proteomics, 2006, 6(13): 3792-3800.
[15]LiuXH,WuXD,HanY,etal.SignalpathwayofcardioprotectioninducedbymonophosphoryllipidAinrabbitmyocardium[J].Pathophysiology, 2002, 8(3):193-196.
[16] Zhang K, Kaufman RJ. Signaling the unfolded protein response from the endoplasmic reticulum[J]. J Biol Chem, 2004, 279(25):25935-25938.
[17]PuthalakathH,O’ReillyLA,GunnP,etal.ERstresstriggersapoptosisbyactivatingBH3-onlyproteinBim[J].Cell, 2007, 129(7):1337-1349.
[18] Zou CG, Cao XZ, Zhao YS, et al. The molecular mechanism of endoplasmic reticulum stress-induced apoptosis in PC-12 neuronal cells: the protective effect of insulin-like growth factor I[J]. Endocrinology, 2009, 150(1):277-285.
[19]KimDS,KwonDY,KimMS,etal.Theinvolvementofendoplasmicreticulumstressinflavonoid-inducedprotectiononcardiaccelldeathcausedbyischaemia/reperfusion[J].PharmPharmacol, 2010, 62(2):197-204.
[20] Liu XH, Zhang ZY, Sun S, et al. Ischemic postconditioning protects myocardium from ischemia/reperfusion injury through attenuating endoplasmic reticulum stress[J]. Shock, 2008, 30(4):422-427.
[21]ZhangZY,LiuXH,HuWC,etal.Caleineurin-myocyteenhancerfactor2cpathwaymediatescardiachypertrophyinducedbyendoplasmicreticulumstressinneonatalratcardiomyocytes[J].AmJPhysiolHeartCircPhysiol, 2010, 298(5):H1499-H1509.
[22] 曹 洁, 杨朝霞, 沈 薇, 等. 靶向PERK基因的shRNA真核表达载体的构建及对内质网应激状态下L02肝细胞凋亡的影响[J]. 中国病理生理杂志, 2011, 27(12):2376-2381.
[23]SzegezdiE,HerbertKR,KavanaghET,etal.Nervegrowthfactorblocksthapsigargin-inducedapoptosisatthelevelofthemitochondrionviaregulationofBim[J].JCellMolMed, 2008, 12(6A):2482-2496.
[24] Chitnis NS, Pytel D, Bobrovnikova-Marjon E, et al. miR-211 is a prosurvival microRNA that regulateschopexpression in a PERK-dependent manner[J]. Mol Cell, 2012, 48(9): 353-364.
猜你喜欢
波谱学杂志(2022年1期)2022-03-15
中学生物学(2021年8期)2021-11-02
现代临床医学(2021年1期)2021-01-26
山东医药(2021年28期)2021-01-11
天津医科大学学报(2019年6期)2019-08-13
中国果业信息(2019年1期)2019-01-05
中国畜牧兽医文摘(2018年6期)2018-07-28
分析化学(2017年12期)2017-12-25
安徽医科大学学报(2015年9期)2015-12-16
科学启蒙(2015年8期)2015-08-07
