
NF-κB结合元件缺失对NOX1启动子转录活性的影响*
2014-08-13 12:06吴炜景徐采云黄文杰
中国病理生理杂志 2014年10期
吴炜景, 李 理, 徐采云, 黄文杰△
(1福建医科大学附属第二医院呼吸内科,福建 泉州 362000; 2广州军区广州总医院呼吸内科,广东 广州 510010)
烟酰胺腺嘌呤二核苷酸磷酸氧化酶(NADPHoxidase,NOX/DUOX)家族是产生ROS的重要来源,是急性肺损伤过程中诱导产生细胞氧化应激的主要参与者。本实验室的前期研究表明,采用代表性的炎症因子TNF-α刺激肺泡Ⅱ型上皮细胞来源的A549细胞,可观察到放大的炎症反应和过度的氧化应激,造成细胞凋亡水平上升,细胞存活率下降[1-2]。因此,明确NOX/DUOX家族基因的表达机制,是炎症反应时调控其衍生的ROS维持在适当水平、减轻内源性氧化损伤的基础。在以高氧诱导的急性肺损伤小鼠模型中,NOX1在肺泡上皮细胞氧化损伤中的作用可能大于NOX2[3]。我们前期的研究提示NF-κB可能参与了TNF-α诱导的肺泡上皮细胞氧化损伤[2, 4],我们推测其与NOX1的转录调控密切相关。鉴此,本研究采用生物信息学分析技术,获取NOX1基因近端启动子区NF-κB结合元件序列,通过构建相应的含NOX1启动子区及NF-κB结合元件缺失的启动子区萤光素酶重组载体,探讨该NF-κB对NOX1基因转录的调控作用。
材 料 和 方 法
1 细胞培养
A549细胞由广州军区广州总医院医学实验科细胞库提供,在含10%灭活小牛血清、1×105U/L青霉素和100mg/L链霉素的RPMI-1640培养基中置于37 ℃、5% CO2、饱和湿度条件下的培养箱中培养,2~3 d传代1次,取处于对数生长期的细胞用于实验。
2 主要试剂
RPMI-1640高糖培养基、胰酶和小牛血清购自HyClone;重组人TNF-α购自PeproTech; Lipofectamine 2000和Opti优化培养基购自Invitrogen;PCR试剂购自康为世纪公司;PCR引物由英骏公司合成;核酸marker DL2000、marker DL10000、琼脂糖、DNA提取试剂盒、胶回收试剂盒和质粒抽提试剂盒购自宝生物公司;pGL3萤光素酶报告基因载体、pRL-TK萤光素酶报告基因载体、双萤光素酶报告基因检测系统、T4 DNA ligase、KpnⅠ和HindⅢ购自Promega;大肠杆菌DH5α 由广州军区总医院医学实验科提供;胰化蛋白胨、酵母提取物和氨苄青霉素购自Sigma;DNA测序由华大基因研究院完成。
3 主要方法
3.1生物信息学分析 根据GenBand提供的NOX1基因DNA序列的CDS(GenBank Accession: NG_012567.1/GI:254939587),确定翻译起始位点为ATG,将该位点定义为+1,提取其5’上游约1 500 bp的序列。登录http://www.gene-regulation.com,采用Alibaba 2.1软件分析上述序列,NOX1近端启动子区约1 500 bp范围内可预测到2个NF-κB结合元件。元件1称为NOX1/NF-κB1,位于-1 095~-1 086,核心序列为5’-CAGGAAAAC-3’;元件2称为NOX1/NF-κB2:位于-261~-252,核心序列为5’-TAAAATCCCC-3’,见图1。我们运用EMSA证实NOX1/NF-κB1不具有与TNF-α探针的结合活性(结果未列),故将翻译起始位点上游-236至-323的序列作为缺失位点。

Figure 1. Identification of NF-κB responsive cis-acting elements in the proximal promoter of human NOX1 gene.
3.2重组载体构建 (1) 采用Primer Premier 5.0软件设计引物,用于扩增NOX1基因近端启动子区全段约1 415 bp,上游和下游引物分别引入KpnⅠ和HindⅢ酶切位点,序列及产物长度见表1,横线部分为KpnⅠ和HindⅢ酶切位点。(2) 采用上述软件设计用于扩增NOX1基因近端启动子区A段和B段的特异性引物,分别称为AF、AR和BF、BR。AF和BR端分别引入KpnⅠ和HindⅢ酶切位点,AR和BF含有重叠部分可将A段和B段连接,产物约1 327 bp,较全长片段缺少-236至-323的序列。引物序列及产物长度见表1。(3) PCR扩增NOX1基因近端启动子区全段、A段和B段: 按试剂盒说明书提取A549细胞基因组DNA,超微量分光光度计NanoDrop定量。反应体系:2×PCR Pre-Master Buffer 10 μL,Forward Primer 0.5 μL,Reverse Primer 0.5 μL,Template DNA 3.0 μL,dH2O 6.0 μL,total 20.0 μL。反应条件:95 ℃ 30 s,95 ℃ 30 s,51 ℃ 30 s,72 ℃ 60 s,共40个循环,72 ℃ 10 min。(4)重叠PCR连接NOX1基因近端启动子区A段和B段并扩增A+B段:按试剂盒说明书将上述A段和B段的PCR扩增产物(分别为NOX1-A和NOX1-B)依次行电泳、回收、纯化。连接A段和B段的反应体系:2×PCR Pre-Master Buffer 10.0 μL,NOX1-A 1.5 μL,NOX1-B 1.5 μL,dH2O 12.0 μL,total 25.0 μL。反应条件:95 ℃ 30 s,95 ℃ 30 s,51 ℃ 30 s,72 ℃ 60 s,共5个循环,72 ℃ 2 min,4 ℃ 10 min。扩增A+B段的反应体系:在上述反应产物中加入2×PCR Pre-Master Buffer 10.0 μL,Primer AF 0.5 μL,Primer BR 0.5 μL,dH2O 14.0 μL,总体系至50.0 μL。反应条件:95 ℃ 30 s,95 ℃ 30 s,51 ℃ 30 s,72 ℃ 60 s,共35个循环,72 ℃ 10 min。(5) 含NOX1近端启动子区的重组pGL3载体的构建:将NOX1基因近端启动子区全片段、A+B片段和pGL3空载体进行KpnⅠ和HindⅢ位点的双酶切、电泳及回收,获取纯化的目的片段和pGL3线性载体,分光光度计定量。以T4 DNA连接酶将目的片段与pGL3线性载体连接,将重组质粒转入大肠杆菌感受态细胞,涂板培养,摇菌并抽提质粒。将质粒进行双酶切、电泳初步鉴定,同时送华大基因研究院DNA测序、比对。比对正确的质粒分别命名为“全长载体pGL3-NOX1-1415”和 “缺失载体pGL3-NOX1-1327”。
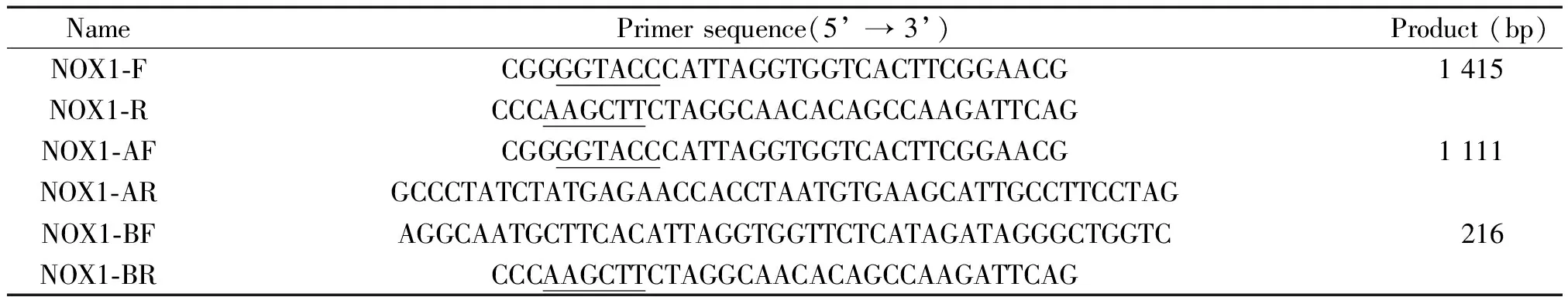
表1 引物序列
3.3细胞萤光素酶活性测定 按照下法配制转染液:取pGL3重组质粒4.0μg和pRL-TK0.4μg加入到250μL的Opti优化培养基,室温静置5min;取Lipofectamine2000 10μL加入到250μLOpti优化培养基,室温静置5min;两者混匀,室温静置20min,用完全培养基稀释至1mL/well后再次静置20min即可用于转染实验。
将细胞分为a组:转染pGL3空质粒;b组:转染pGL3-NOX1-1415质粒;c组:转染pGL3-NOX1-1327质粒;将细胞以2×108/L接种于6孔板,待贴壁细胞的融合度达到50%,倾去培养基,将上述转染液加入细胞培养孔,继续培养6h后停止转染,倾去转染液,以PBS轻轻洗涤3次,瞬时转染结束,继续以含TNF-α(10μg/L)的无血清培养基刺激24h,收获细胞。按试剂盒说明书裂解细胞,Biocell多功能酶标仪先后测定萤火虫萤光素酶活性(记为A1)和海肾萤光素酶活性(记为A2),A1/A2即为萤光素酶活性,表示插入启动子的活性。
4 统计学处理
应用SPSS 13.0软件包分析。数据以均数±标准差(mean±SD)表示,所有数据均进行方差齐性检验。采用单因素方差分析进行组间均数比较,满足方差齐性的多重比较采用LSD法。以P<0.05为差异有统计学意义。
结 果
1 重组质粒pGL3-NOX1-1415和pGL3-NOX1-1327的双酶切电泳结果
各片段PCR产物电泳结果如图2所示:a泳道为片段A,约1 100 bp;b泳道为片段B,约250 bp,c泳道为片段A+B,约1 300 bp;d泳道为全长片段,约1 400 bp。缺失片段A+B较全长片段电泳速度快,提示分子量稍小,缺失成功。

Figure 2. Electrophoresis image of different PCR products. Marker: DL2000; a: fragment A; b: fragment B; c: fragment A+B; d: NOX1 promoter.
全长载体pGL3-NOX1-1415双酶切产物电泳结果如图3所示:a泳道为全长片段;b泳道为空质粒pGL3;c泳道为质粒pGL3-NOX1-1415;d泳道为双酶切产物。位于泳道d的2条酶切产物分别与全长片段、空质粒pGL3处于同一水平,提示该载体确实由两者重组而成。

Figure 3. Electrophoresis image of pGL3-NOX1-1415. Marker: DL10000; a: NOX1 promoter; b: pGL3; c: pGL3-NOX1-1415; d: products of double digestion.
缺失载体pGL3-NOX1-1327双酶切产物电泳结果如图4所示:a泳道为缺失片段A+B;b泳道为空质粒pGL3;c泳道为质粒pGL3-NOX1-1327;d泳道为双酶切产物。位于泳道d的2条酶切产物分别与缺失片段A+B、空质粒pGL3处于同一水平,提示该载体确实由两者重组而成。
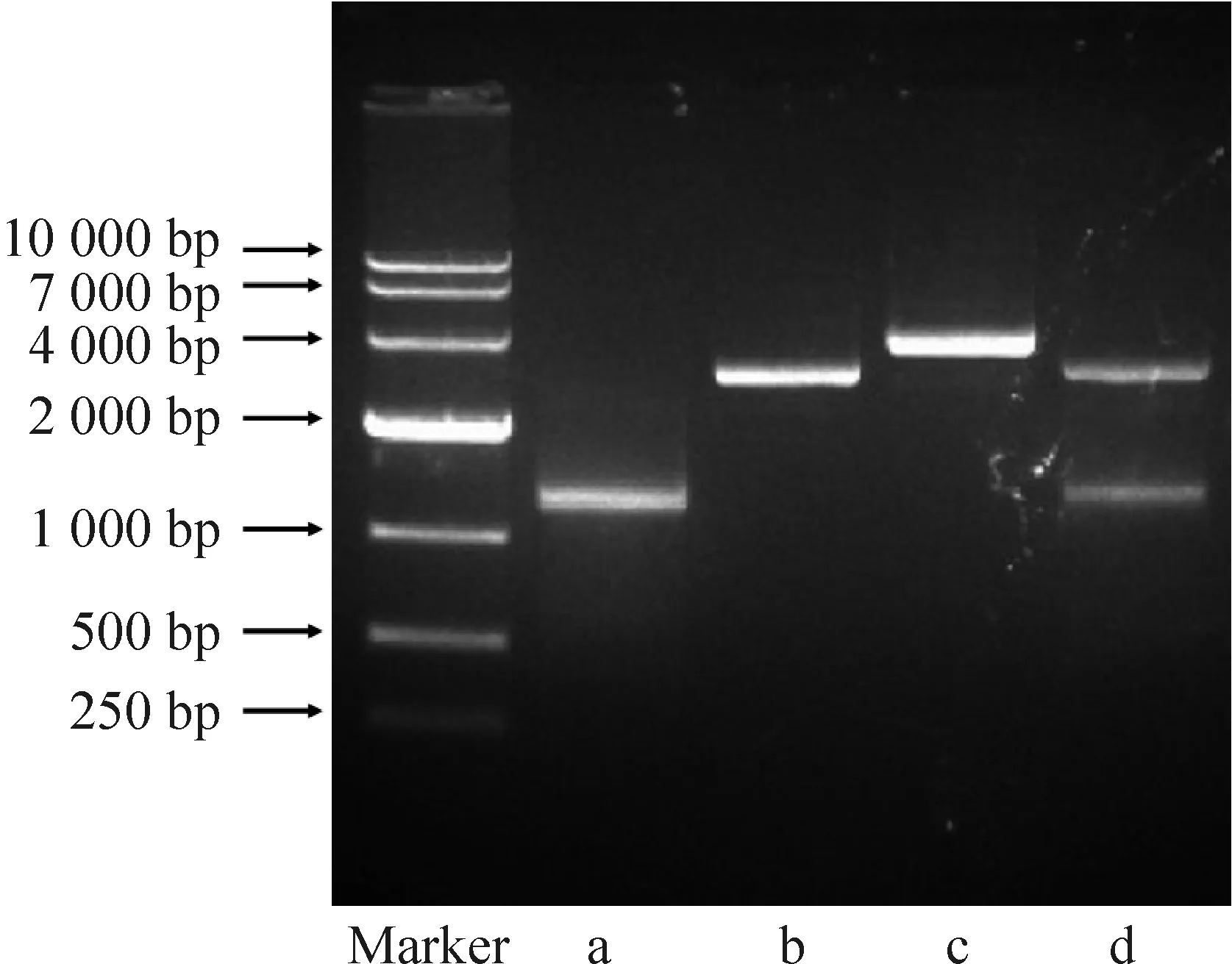
Figure 4. Electrophoresis image of pGL3-NOX1-1327. Marker: DL10000; a: fragment A+B; b: pGL3; c: pGL3-NOX1-1327; d: products of double digestion.
2 重组质粒pGL3-NOX1-1415和pGL3-NOX1-1327的测序结果的对比
2个重组质粒的测序结果分别经NCBI数据库比对,序列正确,载体构建成功。图5所示为核心序列对比,图5A为缺失载体pGL3-NOX1-1327,图5B为全长载体pGL3-NOX1-1415,蓝线所示为两者共有序列,红线所示即为缺失片段,图5A不含该序列,NF-κB结合元件缺失成功。

Figure 5. Sequence comparison of pGL3-NOX1-1415 and pGL3-NOX1-1327. A: pGL3-NOX1-1327; B: pGL3-NOX1-1415. The blue underline shows the same sequence of these 2 vectors. The red underline shows the deleted sequence.
3 细胞萤光素酶活性的比较
单因素方差分析结果显示,分别转染pGL3空质粒、pGL3-NOX1-1415质粒和pGL3-NOX1-1327质粒后,经TNF-α刺激的A549细胞萤光素酶活性的差异有统计学意义(P<0.01)。转染pGL3-NOX1-1415质粒和pGL3-NOX1-1327质粒的A549细胞萤光素酶活性较转染空质粒的A549细胞增强(85.500±3.619vs20.500±1.643, 61.500±3.782vs20.500±1.643),差异有统计学意义(P<0.05)。转染缺失NF-κB结合元件的pGL3-NOX1-1327质粒细胞萤光素酶活性较转染pGL3-NOX1-1415质粒明显降低,差异有统计学意义(P<0.05),见图6。

Figure 6. The luciferase activity of the cells transfected with different plasmids.A: pGL3 group; B: pGL3-NOX1-1415 group; C: pGL3-NOX1-1327 group. Mean±SD.n=6. *P<0.05 vs A; #P<0.05 vs B.
讨 论
炎症反应及其诱发的氧化应激是ALI/ARDS重要的发病机制,针对ALI患者的肺组织保护策略,进行有效抗感染治疗的同时对机体免疫系统合理干预,将过度的炎症反应和氧化应激维持在适当水平,对于保持肺结构细胞完整性同等重要。明确氧化应激相关基因的表达机制,找到其调控靶点从而调节ALI氧化应激损伤,可能比单一补充细胞因子拮抗剂或抗氧化剂效果更好。
NOX1基因是NOX/DUOX家族的重要成员,是调控ALI时上皮细胞ROS生成的主要基因。本实验室前期研究中,以TNF-α刺激肺泡II型上皮细胞A549,观察到细胞上清液中促炎细胞因子IL-1β、IL-6、IL-8及抑炎细胞因子IL-4、IL-10表达水平均上调,氧化物质活性氧、丙二醛生成增加,抗氧化物质超氧化物歧化酶、总谷胱甘肽消耗增多,提示炎症反应放大的同时诱发过度的氧化应激,且存在细胞凋亡水平上调的现象[1-2]。上述实验过程较好地模拟了急性肺损伤发生时II型上皮细胞的炎症环境。在该模型上,我们发现经TNF-α诱导的A549细胞NOX1基因与NF-κBp65基因表达上调趋势相同,提示NF-κB可能参与急性肺损伤过程中NOX1的转录调控。Lee等[5]指出小鼠巨噬细胞NOX1启动子区含有NF-κB结合位点,NF-κB可能是NOX1表达的关键转录因子,其调控作用具有细胞特异性。本研究进一步采用常用的生物信息学软件分析获得NOX1基因近端启动子的2个NF-κB结合元件,该结果与Manea等[6]预测的位点相似。该团队采用CHIP、荧光载体构建等技术证实,在TNF-α刺激的动脉平滑肌上皮细胞中,NF-κB与NOX1基因翻译起始位点上游-258~267区域结合,活化NOX1基因的转录。因此,本实验将包含该核心位点在内的翻译起始位点上游-236~-323区域作为缺失目的序列。
报告基因是用于研究启动子与转录因子相互作用的常用工具。pGL3质粒带有萤火虫萤光素酶的编码区,但不含启动子和增强子,将目的启动子片段插入该质粒,活化的转录因子与该启动子区结合可编码萤光素酶生成,通过检测萤光素酶的活性,可间接反应转录因子对该启动子的转录活性。本研究克隆NOX1近段启动子区约1 400 bp并插入该载体,构建含NOX1基因启动子全长片段的质粒pGL3-NOX1-1415,转染进入A549细胞后经TNF-α刺激24 h,可观察到其萤光素酶活性较转染空质粒显著升高,提示TNF-α刺激后NOX1基因表达明显增强。
NF-κB作为一种具有多向转录调节作用的核因子,广泛参与免疫、炎症、氧化应激、细胞增殖、细胞凋亡等生理病理过程,也是急性肺损伤发生、发展过程中炎症-氧化应激环路中重要的调控节点。TNF-α是ALI过程出现的促炎症细胞因子之一,能促发“瀑布式炎症级联反应”,活化其它细胞因子的释放过程[7]。研究表明,TNF-α可以通过与细胞膜上特异性受体TNFR1结合,激活“RIP1-MEKK3-TAK1-IKK”通路,使胞浆中无活性的NF-κB/I-κB二聚体解离;或者通过与TNFR2结合,募集可降解I-κB的细胞凋亡抑制因子cIAP-1 和cIAP-2,活化NF-κB的核转位。活化后的NF-κB在细胞核内与靶基因的启动子结合,调控其转录[8]。NF-κB的靶基因包括NOX2[9]、诱导型一氧化氮合酶[10]、环氧化酶2[11]、超氧化物歧化酶[12]、谷胱甘肽S转移酶[13]等促氧化和抗氧化相关基因。本研究预测NOX1为NF-κB的靶基因,并以TNF-α作为独立因素刺激A549细胞,模拟ALI诱发的细胞炎症-氧化损伤,将NF-κB结合元件的位置明确为翻译起始位点上游-236至-323的区域,核心序列位于-261 至 -252,为5’-TAAAATCCCC-3’。我们采用重叠延伸PCR技术,成功获得含NOX1近端启动子区部分片段缺失的“缺失载体”pGL3-NOX1-1327。同时荧光素酶检测结果显示,载体pGL3-NOX1-1327启动子区活性较pGL3-NOX1-1415明显下调,这提示-236~323区域(NF-κB结合元件所在区域)含有可被转录因子结合并活化的有效元件;缺少该片段的NOX1基因启动子活性降低。
综上,本研究应用载体构建技术和重叠延伸PCR技术,成功构建含NOX1启动子全长片段的pGL3-NOX1-1415及缺失了该启动子区的有活性NF-κB结合元件的pGL3-NOX1-1327,为后续NOX1基因启动子区的研究奠定了分子生物学基础。缺失NF-κB元件的NOX1基因启动子区转录活性下调,提示在TNF-α诱导肺泡上皮细胞炎症反应继发的氧化应激损伤中,NF-κB参与了NOX1基因转录调控,其具体的结合位点及活性有待后续的实验中运用EMSA、ChIP等技术,从蛋白-DNA结合的角度进一步验证。探讨NF-κB对NOX1的调控机制有助于我们了解急性肺损伤发生时以TNF-α为代表的炎症因子诱发的结构细胞氧化应激损伤机制,为急性肺损伤在基因层面的诊治研究奠定基础。
[参 考 文 献]
[1] 吴炜景, 李跃飞, 李 理, 等. 沉默NF-κBp65基因下调TNF-α诱导的肺泡上皮细胞的炎症反应[J]. 免疫学杂志, 2012, 28(1):15-19.
[2] 吴炜景, 李 理, 袁伟锋, 等. NF-κB p65基因在TNF-α诱导的肺泡上皮细胞氧化损伤中的作用[J]. 中国呼吸与危重监护杂志, 2012, 11(1):46-51.
[3]CarnesecchiS,DeffertC,PaganoA.NADPHoxidase-1playsacrucialroleinhyperoxia-inducedacutelunginjuryinmice[J].AmJRespirCritCareMed, 2009, 180(10):972-981.
[4] Li L, Wu W, Huang W, et al. NF-κB RNAi decreases the Bax/Bcl-2 ratio and inhibits TNF-α-induced apoptosis in human alveolar epithelial cells[J]. Inflammation Res, 2013, 62(4):387-397.
[5]LeeSH,ParkDW,ParkSC,etal.Calcium-independentphospholipaseA2β-Aktsignalingisinvolvedinlipopolysaccharide-inducedNADPHoxidase1expressionandfoamcellformation[J].JImmunol, 2009, 183(11):7497-7504.
[6] Manea A, Tanase LI, Raicu M, et al. Transcriptional regulation of NADPH oxidase isoforms, Nox1 and Nox4, by nuclear factor-κB in human aortic smooth muscle cells[J]. Biochem Biophy Res Commun, 2010, 396(4):901-907.
[7] 李玉梅,卫洪昌.ALI/ARDS抗炎治疗研究的策略与展望[J]. 中国病理生理杂志, 2009, 25(4):813-816, 825.
[8] Morgan MJ, Liu ZG. Crosstalk of reactive oxygen species and NF-κB signaling[J]. Cell Res, 2011, 21(1):103-115.
[9]AnratherJ,RacchumiG,IadecolaC.NF-kappaBregulatesphagocyticNADPHoxidasebyinducingtheexpressionofgp91phox[J].JBiolChem, 2006, 281(9):5657-5667.
[10] Guo Z, Shao L, Du Q, et al. Identification of a classic cytokine-induced enhancer upstream in the human iNOS promoter[J]. FASEB J, 2007, 21(2):535-542.
[11]DengWG,ZhuY,WuKK.Up-regulationofp300bindingandp50acetylationintumornecrosisfactor-alpha-inducedcyclooxygenase-2promoteractivation[J].JBiolChem, 2003, 278(7): 4770-4777.
[12] Jones PL, Ping D, Boss JM. Tumor necrosis factor alpha and interleukin-1beta regulate the murine manganese superoxide dismutase gene through a complex intronic enhancer involving C/EBP-beta and NF-kappaB[J]. Mol Cell Biol, 1997, 17(12):6970-6981.
[13]XiaC,HuJ,KettererB,etal.TheorganizationofthehumanGSTP1-1genepromoteranditsresponsetoretinoicacidandcellularredoxstatus[J].BiochemJ, 1996, 313(Pt1):155-161.
猜你喜欢
成都医学院学报(2022年4期)2022-08-19
江西农业学报(2021年4期)2021-04-20
数学大王·低年级(2020年8期)2020-08-14
三农资讯半月报(2020年11期)2020-06-21
传奇故事(破茧成蝶)(2018年6期)2018-11-14
凤凰生活(2018年8期)2018-09-03
传奇故事(上旬)(2018年6期)2018-07-11
作文与考试·小学高年级版(2017年13期)2017-07-12
小雪花·初中高分作文(2016年9期)2016-05-14
中国当代医药(2015年9期)2015-03-01
