
NF-κB在糖尿病性神经痛中的作用及机制*
2014-08-13 12:06黄阳亮胡景鑫
中国病理生理杂志 2014年10期
黄阳亮, 钟 祎, 胡景鑫
(1广州医科大学生理教研室, 广东 广州 510182; 2中山大学附属第一医院脊柱外科,广东 广州 510080)
糖尿病性神经痛(diabeticneuropathy,DN)是糖尿病最常见的并发症之一,以肢体末梢的慢性病理性疼痛为主要特点,具体表现为痛觉过敏、痛觉超敏和自发性疼痛[1-2]。目前其机制还不清楚,也无有效的治疗和控制措施。
NF-κB在病理性疼痛的发生发展中起重要作用。各种类型的末梢神经损伤模型大鼠背根神经节(dorsalrootganglion,DRG)神经元中都有NF-κB的激活[3-4];用不同方法抑制NF-κB的活化可以明显阻断神经损伤引起的病理性疼痛[5-9]。然而在糖尿病引起的病理性疼痛中,DRG神经元中NF-κB的作用及其机制还有待深入研究。本研究用腹腔注射链脲霉素(streptozocin,STZ)的方法复制大鼠糖尿病模型,痛行为检测方法筛选出现慢性疼痛的动物,检测糖尿病性神经大鼠DRG中p-NF-κB的表达情况,及NF-κB抑制剂对大鼠痛行为的影响并进一步分析其可能的机制。
材 料 和 方 法
1 材料
1.1动物 Sprague-Dawley雄性大鼠(220~260 g),由广东省实验动物中心提供。动物分笼饲养,自由饮食,室温保持在(24±1) ℃,湿度50%~60%。
1.2主要试剂 STZ (Sigma-Aldrich)溶解于柠檬酸缓冲液(pH 4.5)中,腹腔注射STZ (65 mg/kg)复制大鼠糖尿病模型。NF-κB抑制剂PDTC (Sigma)溶解在0.9% NaCl中配成浓度为5 μmol/L的工作液。免疫组化实验用的Ⅰ抗为兔抗大鼠Nav1.7抗体(Sigma-Aldrich),Ⅱ抗为Cy3标记的驴抗兔IgG (Jackson Immuno Research)。Western blotting实验用的Ⅰ抗为兔抗大鼠p-NF-κB p65 (Ser536) 抗体(Cell Signaling Technology)、兔抗大鼠 NF-κB p65抗体(Cell Signaling Technology)和兔抗大鼠GAPDH抗体(Cell Signaling Technology),Ⅱ抗为辣根过氧化酶(horseradish peroxidase, HRP)偶联的羊抗兔IgG (Santa Cruz)。
2 方法
2.1大鼠糖尿病模型的制作 柠檬酸2.1 g加入100 mL双蒸水中配成A液,柠檬酸钠2.94 g加入100 mL双蒸水中配成B液,A和B液以1∶1体积比混合,配成pH 4.2~4.5的柠檬酸缓冲液。将STZ溶解在上述柠檬酸缓冲液中(6.5%),腹腔注射STZ(65 mg/kg)复制大鼠糖尿病模型。术后第4天开始尾静脉采血测血糖,血糖高于20 mmol/L视为高血糖。
2.2痛行为测试 为了让大鼠熟悉测试环境,消除大鼠心理因素对测试结果的影响,于测试前1周把大鼠放于测试箱内15~20min,隔天1次, 共3次。机械性痛敏测试用vonFreyhair方法。测试箱为透明有机玻璃箱(18cm×25cm×18cm),箱底为金属网制成(网格为0.8cm×0.8cm),通过网孔用vonFreyhair可以对大鼠足底部皮肤施加机械性刺激。刺激后大鼠出现迅速的撤足或舔足视为阳性反应。热超敏用的是平台测试的方法。热源置于玻璃板下方,对准大鼠后肢加热,测量大鼠撤足潜伏期, 每次测量要间隔5min以上,取3次测量结果的平均值。筛选出撤足阈值和撤足潜伏期明显降低(即出现痛觉过敏)的模型动物做后续实验。
2.3免疫组织化学 对照组和实验组大鼠,腹腔注射乌拉坦(1.5 g/kg)麻醉,经主动脉快速灌注4 ℃ 肝素化的生理盐水300 mL,然后用4%多聚甲醛300 mL 灌注30 min,解剖动物取出L4和L5 DRG,再放入4%多聚甲醛溶液中固定3 h,随后转入30%蔗糖中脱水2 d。标本脱水后进行冰冻切片(厚度16 μm, Leica CM3050S)并收集于含有0.01 mol/L PBS的24孔板内,用0.01 mol/L PBS洗3次,每次5 min,然后室温下用封闭液(含3%驴血清的0.3% Triton X-100溶液)作用1 h,以封闭非特异性结合位点。吸去封闭液,加入兔抗大鼠Nav1.7抗体(1∶100),4 ℃摇床上孵育48 h后,吸去Ⅰ抗,用0.01 mol/L PBS洗3次,加入Cy3标记的驴抗兔Ⅱ抗(1∶200),避光室温下作用1 h, 再用0.01 mol/L PBS洗3次,随机挑选切片贴于载玻片上,立即于荧光显微镜(Olympus IX71)下观察并拍照。
2.4Westernblotting分离双侧L4~L5DRG组织,在培养皿中去除软脊膜和残留血块,将组织迅速放入液氮罐中(10s), 随后将组织按重量加入Tris裂解缓冲液[超纯水 2.8mL, 1.0mol/LTris(pH6.8) 1mL, 10%SDS6mL, 2%β-巯基乙醇 0.2mL; 10mL配方]中,加入蛋白酶抑制剂混合物cocktail(1∶1 000;RocheMolecularBiochemicals) 和磷酸酶抑制剂 (1∶1 000), 冰上匀浆,超声破碎, 低温离心。 取上层含组织蛋白的液体分装保存在-80 ℃。用Micro-BAC法测定蛋白浓度,取等量蛋白样品(50μg)加入1/10体积的上样缓冲液,混匀沸水浴10min变性。随后加入SDS-PAGE胶孔中,电泳分离蛋白,电转至PVDF膜上。常温下5 %封闭液封闭PVDF膜1h,I抗孵育,4 ℃摇床过夜.洗脱3次,每次15min,常温下II抗孵育2h,洗脱3次,每次15min,ECL显色,曝光。
3 统计学处理
用SPSS 10.0统计软件处理,数据以均数±标准差(mean±SD)表示,用单因素方差分析(ANOVA)或t检验比较组间差异,以P<0.05为差异有统计学意义。
结 果
1 NF-κB在糖尿病性神经痛中的作用
本实验用腹腔注射STZ (65 mg/kg)的方法复制大鼠糖尿病模型。与对照组 (柠檬酸缓冲液溶剂组)相比,STZ组大鼠给药后2周开始血糖明显升高(血糖高于20 mmol/L),持续至给药后12周[6周血糖值:(25.40±6.32) mmol/L;12周血糖值:(22.80±5.24) mmol/L]。痛行为学测试方法检测大鼠双侧后肢撤足阈值(paw withdrawal threshold, PWT)和撤足潜伏期(paw withdrawal latency, PWL),与对照组相比,STZ组大鼠给药后4周开始出现PWT和PWL明显下降,持续至给药后12周,见图1。连续7 d鞘内注射NF-κB抑制剂PDTC (5 μmol/L,每次15 μL, 每天1次;STZ给药前1 h开始)可以明显提高大鼠的PWT和PWL,大鼠慢性疼痛症状明显减轻,见图2。这些结果表明NF-κB的活化可能参与了STZ诱导的慢性糖尿病性神经痛的形成与维持。
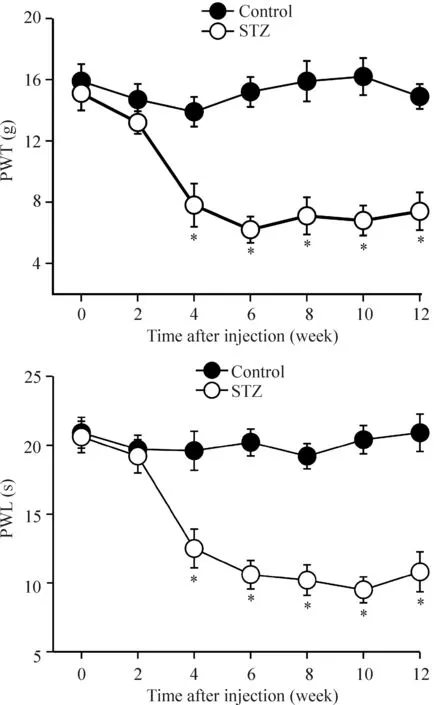
Figure 1. STZ induced mechanical allodynia and thermal hyperalgesia.Mean±SD. n=8. *P<0.05 vs control group.
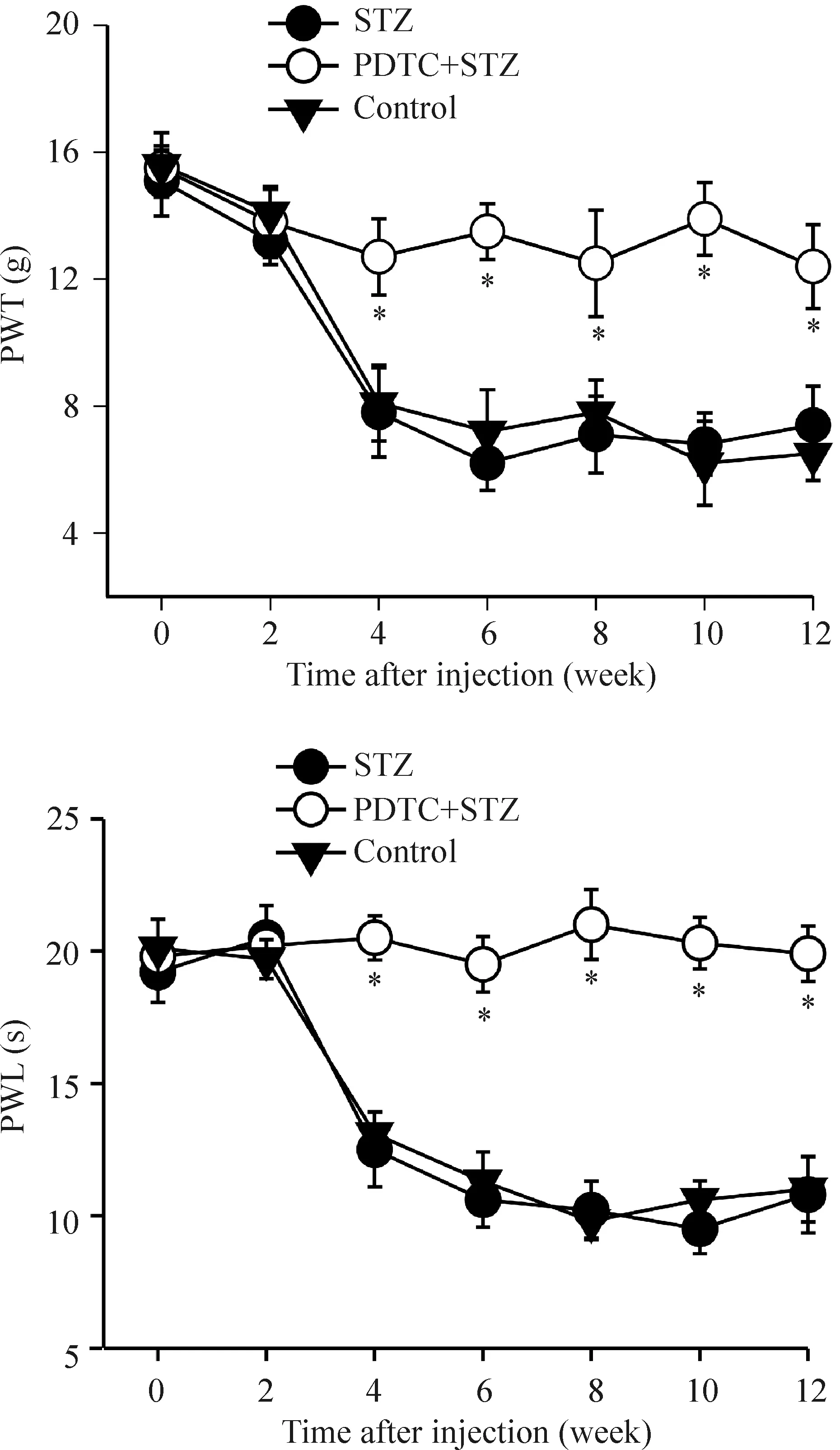
Figure 2. Blockade of NF-κB by PDTC attenuated mechanical allodynia and thermal hyperalgesia.PDTC, a potent inhibitor of NF-κB at a concentration of 5 μmol/L (in a volume of 15 μL), was injected intrathecally 1 h before STZ and then daily for 7 days. Control group received solvent.Mean±SD.n=8. *P<0.05 vs STZ group.
2 PDTC明显抑制STZ引起的p-NF-κB表达上调
与对照组相比,STZ组大鼠给药后6周,L4、L5 DRG中p-NF-κB表达明显升高,t-NF-κB总量没有明显变化。鞘内注射NF-κB抑制剂PDTC可以明显降低STZ引起的p-NF-κB表达上调,且对t-NF-κB无显著影响,见图3。

Figure 3. PDTC attenuates up-regulation of p-NF-κB expression in DRG induced by STZ after 6 weeks.Mean±SD.n=5.*P<0.05 vs control group; #P<0.05 vs STZ group.
3 鞘内注射PDTC明显抑制STZ引起的大鼠L4和L5 DRG中Nav1.7表达上调
我们之前的研究发现糖尿病性神经痛大鼠DRG神经元中电压门控性钠离子通道Nav1.7表达明显升高,时程和疼痛产生维持时间一致[10]。然而钠通道上调的原因尚不清楚。本研究发现抑制NF-κB的活化可以明显降低STZ引起的Nav1.7表达上调,见图4。这表明NF-κB激活后可能通过上调Nav1.7的表达和功能,引起神经元兴奋性增加,异常放电,从而导致慢性疼痛的产生和维持。
讨 论
本实验结果显示,STZ诱导糖尿病后,约半数大鼠会出现慢性疼痛症状。表现为双侧下肢的机械性痛觉过敏和热痛觉超敏,鞘内注射NF-κB 抑制剂PDTC可以明显降低STZ诱导的L4和L5 DRG中NF-κB 磷酸化水平的升高,明显抑制DRG中Nav1.7的表达上调,并且改善大鼠慢性疼痛的症状。因此NF-κB 的活化在糖尿病性神经痛的产生和维持中起重要作用,其机制可能和上调DRG中的钠通道表达和功能有关。
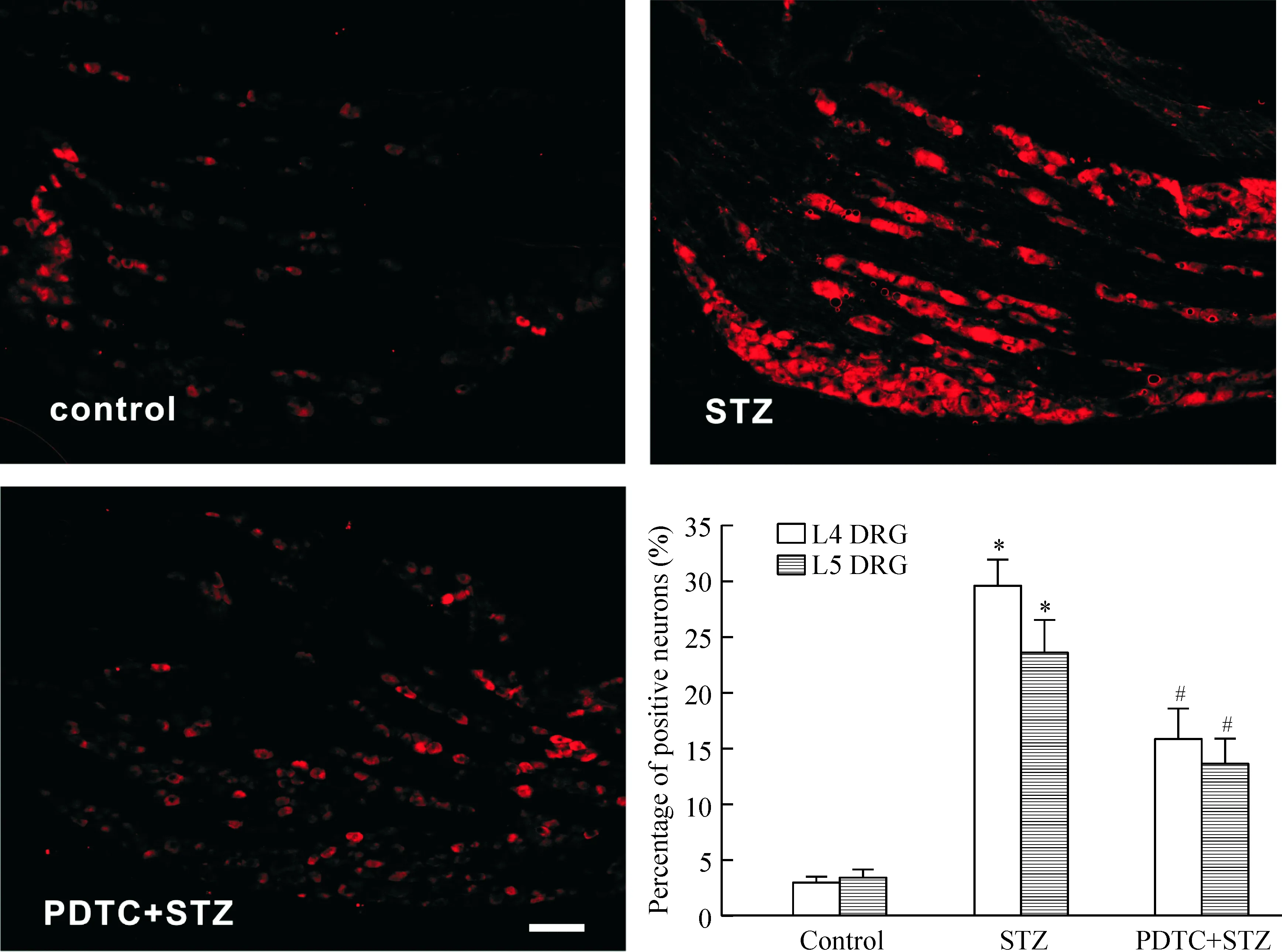
Figure 4. PDTC attenuated up-regulation of Nav1.7 expression in DRG neurons induced by STZ after 6 weeks.Scale bar=100 μm. Mean±SD.n=5.*P<0.05 vs control group; #P<0.05 vs STZ group.
NF-κB在病理性疼痛的发生发展中起重要作用。各种类型的末梢神经损伤模型大鼠DRG神经元中都有NF-κB的激活[3-4];用不同方法抑制NF-κB可以明显阻断病理性疼痛[5-9]。最近有研究发现NF-κB上游激酶抑制性κB激酶(inhibitory kappa B kinase, IKK)的抑制剂可阻断STZ引起的慢性疼痛,同时末梢神经血流和神经传导速度也得到改善[11]。本研究发现鞘内注射PDTC抑制NF-κB可以阻断STZ引起的机械性痛觉过敏、热痛觉超敏、DRG中p-NF-κB 的过表达。这表明IKK-NF-κB信号通路的激活是病理性疼痛产生的重要原因。
NF-κB活化后可以引起许多目的基因的转录[12],并导致病理性疼痛的发生发展。电压门控性钠离子通道Nav1.7属于河豚毒素敏感型(tetrodotoxin-sensitive,TTX-S)钠离子通道,在DRG的感觉神经元上有表达,并且在炎症疼痛和神经病理性疼痛模型动物上表达明显增加[13-14]。高表达Nav1.7的神经元可以表现出明显的异常放电,兴奋性增加[14],然而其机制还不清楚。我们的研究发现NF-κB 的抑制剂可以明显阻断STZ诱导的Nav1.7的表达上调,这表明NF-κB活化是Nav1.7表达上调的重要原因,Nav1.7可能作为NF-κB的下游参与STZ诱导的糖尿病性神经痛。
综合以上分析,我们认为NF-κB的激活在糖尿病性神经痛的发生发展中起重要作用,活化的 NF-κB可能通过上调DRG神经元的电压门控性钠通道,引起神经元自发放电,兴奋性增加,进而诱发糖尿病性神经痛的产生。以NF-κB为靶点的药物将有助于治疗糖尿病性神经痛。
[参 考 文 献]
[1] Calcutt NA, Freshwater JD, Mizisin AP. Prevention of sensory disorders in diabetic Sprague-Dawley rats by aldose reductase inhibition or treatment with ciliary neurotrophic factor [J]. Diabetologia, 2004, 47(4): 718-724.
[2]SerraJ.Microneurography:towardsabiomarkerofspontaneouspain[J].Pain, 2012, 153(10): 1989-1990.
[3] Ma W, Bisby MA. Increased activation of nuclear factor kappa B in rat lumbar dorsal root ganglion neurons following partial sciatic nerve injuries [J]. Brain Res, 1998, 797(2): 243-254.
[4]ZangY,HeXH,XinWJ,etal.InhibitionofNF-κBpreventsmechanicalallodyniainducedbyspinalventralroottransectionandsuppressesthere-expressionofNav1.7inDRGneuronsin vivoandin vitro [J].BrainRes, 2010, 1363: 151-158.
[5] Fu ES, Zhang YP, Sagen J, et al. Transgenic inhibition of glial NF-kappa B reduces pain behavior and inflammation after peripheral nerve injury [J]. Pain, 2010, 148 (3): 509-518.
[6]MeunierA,LatrémolièreA,DominguezE,etal.Lentiviral-mediatedtargetedNF-κBblockadeindorsalspinalcordgliaattenuatessciaticnerveinjury-inducedneuropathicpainintherat[J].MolTher, 2007, 15(4): 687-697.
[7] Niederberger E, Schmidtko A, Gao W, et al. Impaired acute and inflammatory nociception in mice lacking the p50 subunit of NF-κB [J]. Eur J Pharmacol, 2007, 559 (1): 55-60.
[8]SunT,SongWG,FuZJ,etal.Alleviationofneuropathicpainbyintrathecalinjectionofantisenseoligonucleotidestop65subunitofNF-κB[J].BrJAnaesth, 2006, 97 (4): 553-558.
[9] Tegeder I, Niederberger E, Schmidt R, et al. Specific inhibition of IκB kinase reduces hyperalgesia in inflammatory and neuropathic pain models in rats [J]. J Neurosci, 2004, 24 (7): 1637-1645.
[10]HuangYL,ZangY,ZhouLJ,etal.TheroleofTNF-alpha/NF-kappaBpathwayontheup-regulationofvoltage-gatedsodiumchannelNav1.7inDRGneuronsofratswithdiabeticneuropathy[J].NeurochemInt, 2014,75:112-119.
[11] Negi G, Sharma SS. Inhibition of IκB kinase (IKK) protects against peripheral nerve dysfunction of experimental diabetes [J]. Mol Neurobiol, 2014 Jun 20. [Epub ahead of print]
[12]PahlHL.ActivatorsandtargetgenesofRel/NF-κBtranscriptionfactors[J].Oncogene, 1999, 18(49): 6853-6866.
[13] Chattopadhyay M, Mata M, Fink DJ. Continuous delta-opioid receptor activation reduces neuronal voltage-gated sodium channel (NaV1.7) levels through activation of protein kinase C in painful diabetic neuropathy [J]. J Neurosci, 2008, 28(26): 6652-6658.
[14]HongS,MorrowTJ,PaulsonPE,etal.Earlypainfuldiabeticneuropathyisassociatedwithdifferentialchangesintetrodotoxin-sensitiveand-resistantsodiumchannelsindorsalrootganglionneuronsintherat[J].JBiolChem, 2004, 279(28): 29341-29350.
猜你喜欢
保健医苑(2022年5期)2022-06-10
中国典型病例大全(2022年7期)2022-04-22
感染、炎症、修复(2021年1期)2021-07-28
昆明医科大学学报(2021年2期)2021-03-29
昆明医科大学学报(2021年2期)2021-03-29
中国科技纵横(2021年24期)2021-03-02
蚕桑通报(2020年1期)2020-07-10
妇女(2020年4期)2020-04-20
中国土壤与肥料(2018年5期)2018-11-05
中成药(2018年6期)2018-07-11
