
DART-MS/MS快速检测食品中非法添加的N-苄基他达拉非等7种新型PDE-5型抑制剂
2023-08-28 02:22董培智赵丽红
食品与药品 2023年4期
任 静,董培智,赵丽红
(1. 山西中医药大学,山西 晋中 030619;2. 山西省检验检测中心药品检验技术研究所 食品药品安全防控山西省重点实验室,山西 太原 030031)
近年,很多不法商家在利益的驱动下,在一些食品中非法添加化学物质以增强其宣称功能。由于添加的化学物质未经毒理学实验评价且剂量随意,对消费者的身心健康带来一定隐患[1]。因此,解决食品安全中存在的非法添加乱象及建立相应的检测方法显得尤为重要。
目前,文献报道中有关磷酸二酯酶5(PDE-5)抑制剂传统检测方法多为高效液相色谱法[2-4],液相色谱串联质谱法[5-9],核磁共振波谱法[10-12]等。上述方法中,所使用的仪器设备较大,大多需要繁琐的样品提取、制备和分离过程[13],且成本高,污染大。理想的快速检测方法应该是具有快速、省时、简便、环保等特点[14]。
近年兴起的实时直接分析(direct analysis in real time,DART)质谱是一种新型的质谱电离技术[15-16]。其原理是利用He或N2气体分子在高压放电针下形成辉光放电产生等离子体,随后等离子体打在待测品表面,使待测物品分子电离,产生的离子进入质谱检测器进行检测[17-19]。DART质谱不再需要繁琐的样品前处理、制备大量的流动相,只需几秒钟就可分析单个样品,特别适合样品的实时、快速、高通量筛查[20]。
本研究以市售宣称具有抗疲劳等作用的食品为研究对象,采用DART-MS/MS技术对非法添加的PDE-5抑制剂的分析条件进行优化,建立了一种高效、准确、快速、简单的分析方法,以期为保障食品质量安全提供技术支撑。
1 仪器与材料
1.1 仪器
Agilent Ultivo LC/TQ质谱仪(美国安捷伦公司);SVP-DART离子源(美国IonSense公司);Eppendorf移液器(德国艾本德公司);DV215CD电子天平(美国奥豪斯公司)。
1.2 药品与试剂
3-羟丙基去甲他达拉非对照品,羟基卡巴地那非对照品,环己基去甲他达拉非对照品,四氢咔啉对照品,N-苯基丙氧苯基卡巴地那非对照品,N-苄基他达拉非对照品(加拿大TLC Pharmachem公司);丙氧苯基去碳去甲基卡巴地那非对照品(湖北省药品监督检验研究院)。以上对照品纯度均≥96.0 %。乙腈(色谱纯,美国迪马公司);高纯氦气,氮气(纯度≥99.999 %,太原市泰能气体有限公司)。牡蛎粉,蛋白粉,咖啡,金咖啡,虫草强肾片样品为历年抽检样品;海参肽片、鹿鞭牡蛎粉由上海市食品药品检验研究院提供。
2 方法
2.1 对照品溶液的配制
分别准确称取7种那非类对照品各5 mg,置入50 ml量瓶,用乙腈溶解并定容,得到100 μg/ml的储备溶液,保存在0~5 ℃冰箱中,临用前用乙腈稀释为10 μg/ml的工作液或配制成10 μg/ml的混合溶液。
2.2 样品前处理
取样品适量,混匀,研细,取约2 g,精密称定,置入50 ml量瓶,用乙腈定容[21],然后手动振摇混合溶液10 s。用移液器吸取4 μl溶液置玻棒尖端,滑动速度为0.2 mm/s,然后在氦气流中放置约10 s,进样。
2.3 检测条件
DART条件:氦气为离子化气体,氮气为待机气体,采用12位液体样品进样模块(12-Dip-it Module)进样,筛网电压为250 V,离子源出口距质谱进口为2.5 cm,气体压力为0.50 MPa,用浓度为10μg/ml的单一对照品溶液和10 μg/ml的混合对照品溶液分别优化7种PDE-5抑制剂的离子源温度、进样速率和进样体积,以碎裂离子的总峰面积选择最优参数,其余DART参数均为仪器默认设置。
MS/MS条件:正离子模式采集;碎裂电压135 V;质量扫描范围为m/z100~1000;采用多反应监测(MRM)模式,将浓度为10 μg/ml的7种PDE-5标准溶液进行二级质谱碎裂,以碎裂离子峰的峰面积选定最优碰撞能量(见表1),其余质谱参数为仪器默认参数。
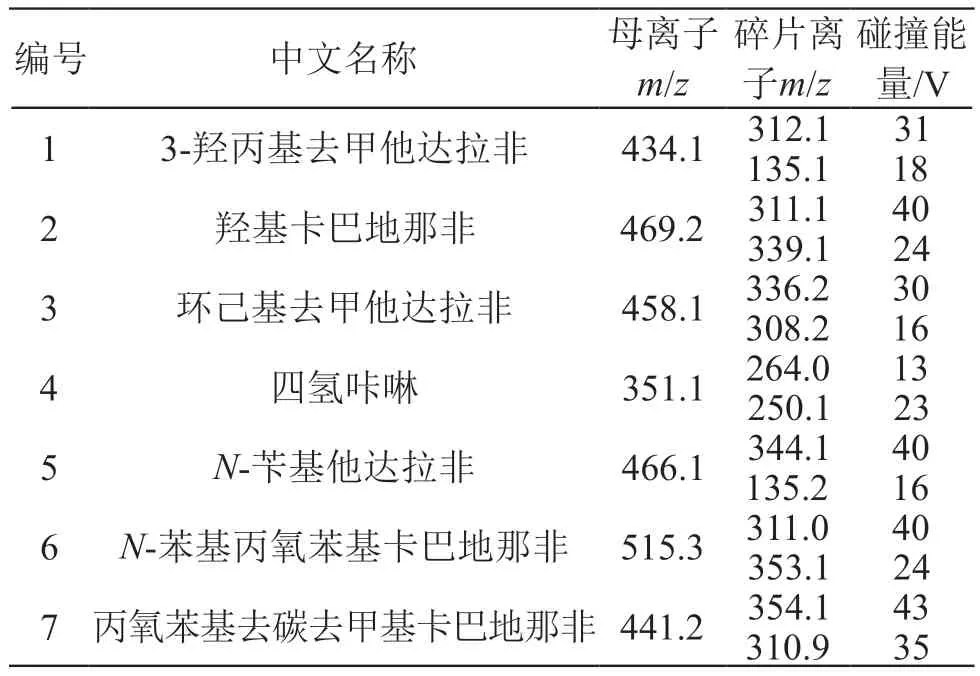
表1 7种PDE-5抑制剂的质谱检测参数
2.4 定性过程
用移液器吸取4 μl混合对照品溶液(10 μg/ml)点于自动进样架上的玻棒尖端,DART离子源和MS/MS条件见2.3,在MRM模式下进行二级质谱碎裂,确定了7种PDE-5化合物的碎片离子峰(见表1),并利用离子丰度比进行定性分析,当样品色谱图中所选择监测离子对的相对丰度比与相当浓度的对照品溶液的离子相对丰度比(K)偏差不超过规定范围[21],即可判定样品中检出相应的化合物。
3 结果与分析
3.1 DART离子源条件的优化
3.1.1 离子源温度的选择 对于DART离子源,其温度的改变会影响化合物的离子化效率。DART加热室的温度提高,激发态氦原子的离子化能力也随之增强,使得分子离子碎片的比例提高[22]。因此,实验中用单一对照品溶液对DART离子源温度进行优化。分别取适量10 μg/ml单一对照品溶液,分别于300~500 ℃(以50 ℃为梯度)下进样,利用每个温度下测得的离子总峰面积来评价离子源温度对检测灵敏度的影响,结果见图1。由图1可见,温度在300~500 ℃范围内,离子化效率随温度的升高呈上升趋势。300 ℃时7种化合物的总峰面积很小,这可能是由于较低温度下分析物的解吸不良导致[23]。超过450 ℃后离子信号减弱,这可能是由于温度过高使得样品分解、碳化[24]。综合考虑最终选择的最佳温度为450 ℃。
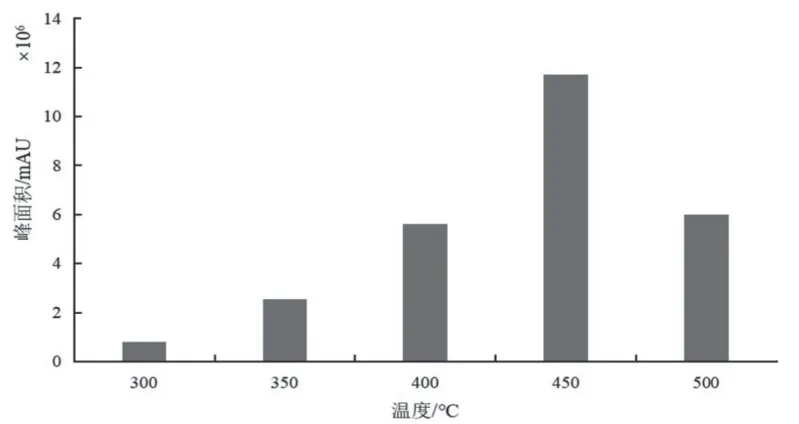
图1 7种PDE-5抑制剂离子总峰面积与离子源温度的关系
3.1.2 样品传输速度的选择 对于DART离子源,其软件操控的滑动轨道速度也会对结果造成影响。一般来说样品传输速度越小,离子化气体与目标化合物接触越充分,进入检测器的离子越多,离子响应强度也越高,反之,离子化强度越低[25]。因此,本试验用混标溶液对样品传输速度进行优化。分别取适量10μg/ml混合对照品溶液,分别在0.2~1.0 mm/s(以0.2 mm/s为梯度)速度下进样,利用每个速度下测得的离子总峰面积来评价样品传输速度对离子化效率的影响,结果见图2。由图2可见,样品在0.2 mm/s的传输速度下得到的离子峰面积最大,且峰形良好。综合其对各检测值的影响,最终选择的最佳传输速度为0.2 mm/s。
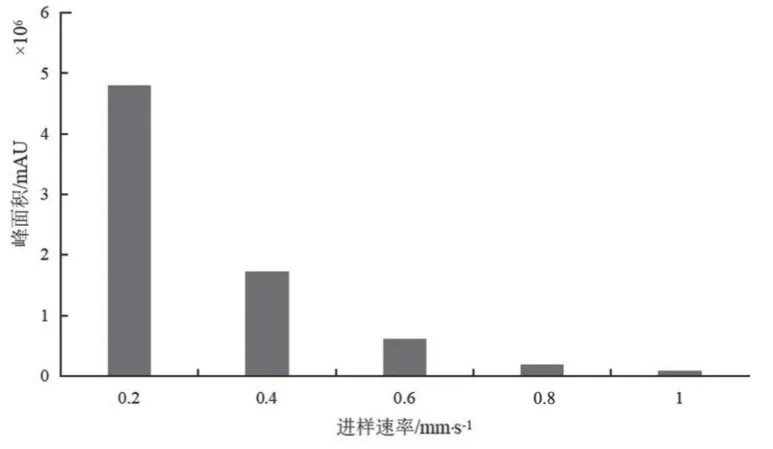
图2 7种PDE-5抑制剂离子总峰面积与进样速率的关系
3.1.3 进样体积的选择 实验中发现利用玻管蘸取混合对照品溶液置氦气流中,其质谱信号弱,且峰形较差。故采用移液器吸取混合对照品溶液置玻管尖端的方式,对进样体积进行优化。分别吸取2~10 μl(以2 μl为梯度)浓度为10μg/ml的混合对照品溶液进样,利用每个待测体积下测得的离子总峰面积来评价进样体积对检测灵敏度的影响,结果见图3。由图3可见,离子信号强度随进样体积的增加而增加,但随着进入质谱仪的离子增多,而质谱仪中可容的离子数量有限,离子的响应强度也会随之减弱[26]。综合考虑,选择4μl作为最优进样量。
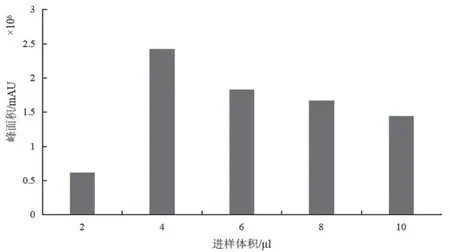
图3 7种PDE-5抑制剂离子总峰面积与进样体积的关系
3.2 专属性
分别取空白溶剂、对照品溶液,并将收集好的阴性、阳性、基质样品按2.2项下方法操作,获得相应色谱图,见图4。结果表明,7种被测成分的响应均未受到溶剂、样品组分的干扰,且响应度良好。
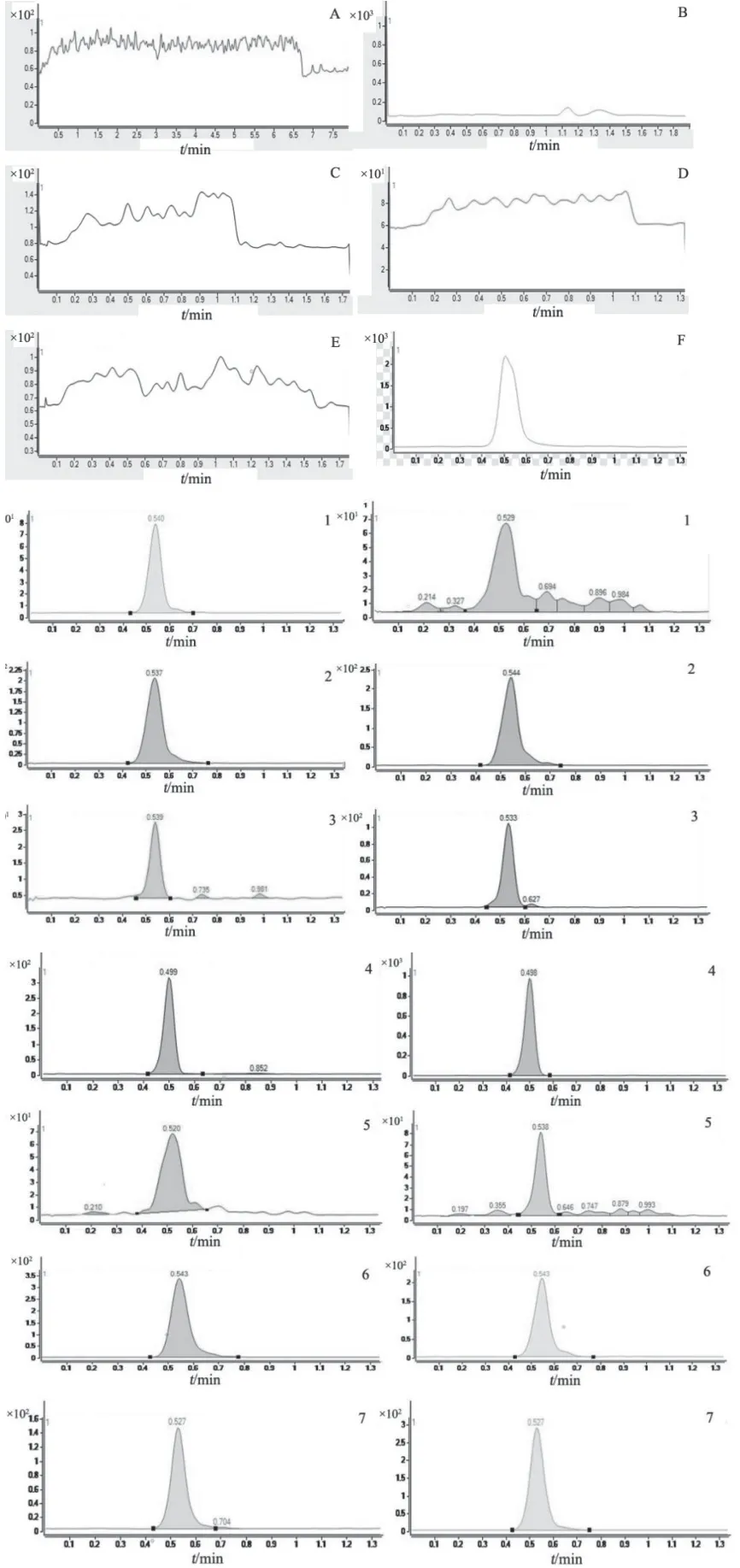
图4 DART-MS/MS检测食品中7种新型PDE-5型抑制剂的典型图谱
3.3 重复性
取阴性蛋白粉样品适量,加入一定体积的对照品溶液,混匀,晾干,自制同一含量的阳性样品粉末6份并进行测定。3-羟丙基去甲他达拉非、羟基卡巴地那非、环己基去甲他达拉非、四氢咔啉、N-苄基他达拉非、N-苯基丙氧苯基卡巴地那非、丙氧苯基去碳去甲基卡巴地那非的平均含量分别为1.36,0.7,12,0.6,6.2,0.6,3.2 μg/g;RSD分别为8.7 %,10.1 %,2.4 %,3.4 %,2.8 %,5.2 %,10.8 %。
3.4 稳定性
分别考察3.3项下自制阳性样品溶液于25 ℃放置0,2,4,8,12,24 h的稳定性,并进样分析。结果表明,自制阳性样品在室温考察时间内均稳定,RSD为1.6 %~6.0 %。
3.5 检出限
在上述优化后条件下,在空白基质中加入混标溶液,样品经前处理后进样检测。以3倍信噪比时进样浓度为检出限确定了7种PDE-5抑制剂的检出限,见表2。由表2可见,该方法PDE-5抑制剂的检出限为0.025~12.5 μg/g,可满足此类药物的筛查要求。
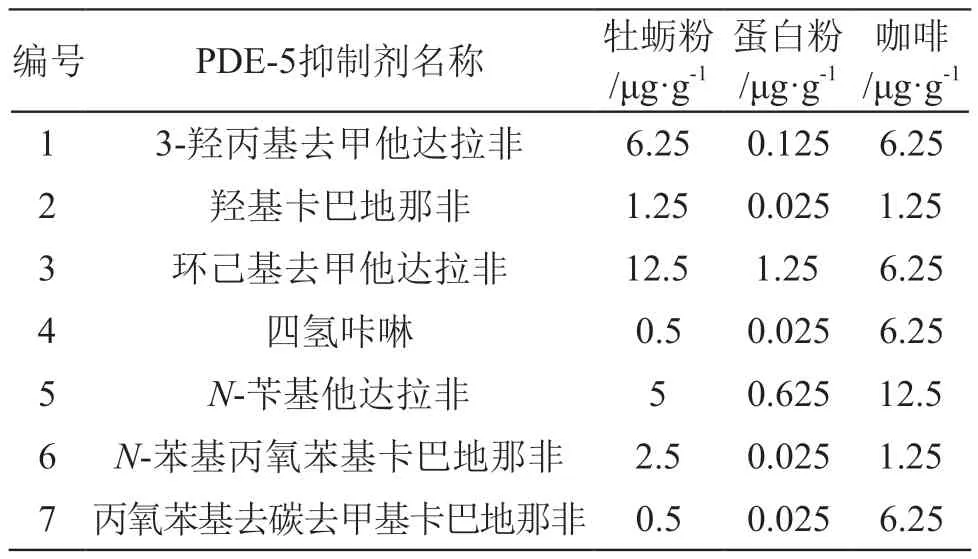
表2 7种PDE-5抑制剂检出限

表3 180批市场抽检样品检出情况
3.6 样品检测
采用本方法对180批次市场抽检样品进行检测。比较样品与对照品的分子离子峰及碎片离子峰的相对离子丰度:样品色谱图中所选择监测的离子相对丰度与相当浓度对照品溶液的离子相对丰度比(K)偏差不超过规定范围[±(20 %~25 %)][21],检测结果发现4种阳性样品,分别非法添加羟基卡巴地那非、环己基去甲他达拉非、N-苄基他达拉非和N-苯基丙氧苯基卡巴地那非。
4 结论
本试验建立了采用DART离子源结合三重四极杆质谱定性检测3种基质中7种PDE-5抑制剂的方法。该方法与传统的液相、液质等技术相比,不需要复杂的样品前处理、耗时的色谱分离及大量化学试剂的消耗,适用于食品中非法添加的PDE-5抑制剂的快速筛查。
猜你喜欢
山西文学(2023年6期)2023-06-09
中成药(2022年10期)2022-12-04
分析化学(2020年8期)2020-08-21
中国酿造(2020年6期)2020-08-02
特产研究(2018年4期)2018-12-11
分析化学(2018年4期)2018-11-02
分析化学(2018年7期)2018-09-17
检验医学与临床(2018年4期)2018-03-06
色谱(2015年6期)2015-12-26
低温与特气(2012年4期)2012-01-10
