
涂膜粉碎法制备原花青素缓释微粒的研究
2023-08-28 02:22彭苑哲王文苹
食品与药品 2023年4期
王 皎,彭苑哲,普 青,王文苹,
(1. 云南中医药大学 中药学院,云南 昆明 650500;2. 云南省高校外用给药系统与制剂技术研究重点实验室,云南 昆明 650500;3. 云南省南药可持续利用重点实验室,云南 昆明 650500)
葡萄籽原花青素(PC)是由不同数量的儿茶素、表儿茶素单体聚合而成的多聚体,具有抗氧化、抗癌、抗菌、抗辐射等药理作用;因结构中富含羟基,故抗氧化活性是维生素C的几十倍,是目前世界上公认的最有效的天然抗氧化剂,已在食品、药品和化妆品领域广泛应用[1-3]。但PC稳定性不佳,易受光照、温度、氧化剂等多重因素影响,且PC在胃肠道中不易吸收,口服生物利用度较低[4],致使其应用受限。
乙基纤维素(ethyl cellulose,EC)已广泛用作药物载体,其不溶于水,但在乙醇等有机溶剂中溶解性较高。EC具有无毒无害、廉价易得、富含羟基、易被改性、可生物降解、可塑性强等优点,可形成机械性能良好的韧性薄膜[5]。EC薄膜具有良好的热稳定性,即使在低温下仍保有一定的柔韧性。此外,还可通过一些简单的方法将EC制备成密度更小、力学性能更好的多孔聚合物微粒,粒径多为微米级,在药物的保存、运输、释放过程中起隔绝污染和破坏的保护作用,同时控制药物释放速度以改善药效[6]。EC在人体胃部酸性环境中也无法溶解,口服后能通过肠道排出,对人体无毒副作用。
微粒是一种常见缓控释药物载体,其容纳范围广、释药行为可调,且能实现药物固态化和稳定化。其一般制法包括乳化-溶剂挥发、反溶剂沉淀、喷雾干燥或高压电纺丝工艺等,需特殊设备[7];另有一种涂膜粉碎法,其操作简单、无需特殊设备和有机溶剂、物料利用率高,目前仅有少数报道涉及。因此本研究以EC为载体材料、PC为模型药物,采用涂膜粉碎法制备载药微粒,并初步考察工艺和药载比例对微粒形态、成型过程、载药量和包封率、相互作用、体外释放等的影响,为涂膜粉碎工艺的深入研究和推广利用提供参考。
1 仪器与试剂
1.1 仪器
BCE224-1CCN十万分之一电子天平(北京赛多利斯);T6新世纪紫外分光光度计(北京普析);Great20傅立叶变换红外光谱仪(天津中科瑞捷);TGL-16.5M高速冷冻离心机(上海卢湘仪);MVE067464-60046-S Phenom飞纳台式扫描电镜(Thermo Fisher scientific);THZ-100恒温培养摇床(上海一恒);SG250HP超声波清洗器(上海冠特);EU-KI-20TF纯水机(南京欧铠);针头式过滤器(常德比克曼)。
1.2 试剂
PC(天津尖峰,批号:002-1905036-10);EC(江西阿尔法高科药业,批号:20220210);4-二甲基氨基肉桂醛(DMAC,上海麦克林);氢氧化钠,磷酸二氢钾(天津风船);盐酸(云南杨林工业开发区汕滇药业);水为超纯水;其余试剂均为分析纯。
2 方法与结果
2.1 PC测定方法
用70 %乙醇配制0.5 mg/ml PC溶液。精密量取1.00 ml PC溶液,用70 %乙醇分别稀释4,5,6,8,10,18,40倍,分别取1 ml各浓度溶液和70 %乙醇空白溶液,加DMAC显色剂[8]3 ml,室温放置30 min,于644 nm处测定吸收值,以PC浓度(mg/ml)为横坐标,吸光度为纵坐标,绘制标准曲线,得回归方程:A=6.4852C+0.0313,r2=0.9991。结果表明,PC在12.5~125 μg/ml浓度范围内线性关系良好。精密度试验RSD为0.39 %,重现性试验RSD为1.75 %,加样回收率在99.46 %~106.29 %范围内,RSD为3.24 %。
2.2 微粒的制备
采用涂膜粉碎法[9]:将PC、EC分别溶于80 %乙醇溶液中,按药载比PC:EC=1:5,2:3,2:1混匀;将混合液涂布于水平洁净玻璃板上,室温下自然挥干,将膜刮下研细过100目筛,即得微粒样品,分别记为PE15、PE23、PE21,密封,置阴凉干燥处保存。同法分别制备空白微粒(EC-MP)和药物微粒(PC-MP)作为对照。
2.3 微观形态
取少量上述样品以双面导电胶固定于样品台上,喷金处理后置于扫描电子显微镜(scanning electron microscope,SEM)下观察其形态。结果见图1。
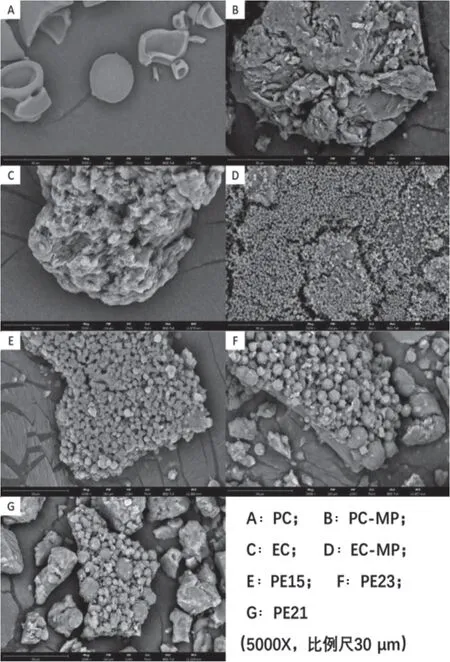
图1 SEM下样品微观形态
由图1可知,PC原料呈皱缩粒子(图1A),个别为饱满球形、表面光滑,粒径约在10~25 µm范围内,大量粒子破裂、暴露出内部空心,壁厚约1~2 µm;经醇溶挥干再粉碎后,PC形态发生显著改变(图1B),呈50~80 µm的疏松类球形微粒,由1~2 µm球形或不规则粒子相互黏接、融合形成。
EC本身为不规则颗粒、粒径较大(60~70 µm),表面粗糙、密布孔道、结构疏松(图1C);经处理后,自发转变为均匀细小的球形微粒,直径约0.7~2 µm、表面光滑,大量粒子密集排列、粘连成片(图1D)。
当药物与辅料混合处置后,不同PC:EC比例所得样品含大量球形粒子、聚集成块片状(图1E、F、G);样品中EC含量越高,则微粒粒径较小且均匀度更佳,其中PE15粒径为2~3 µm,PE21粒径为3~10 µm;随着PC比例增大,逐渐出现一些不规则块状物,其断面略粗糙,推测可能是在溶剂挥发过程中发生了相分离,形成微粒的同时产生块状物。还可能是因EC具有黏性,使载药微粒在干燥过程中形成粘连结块,在一定程度上影响微粒粒径分布。
2.4 微粒形成过程的观测
取少量药物、载体或二者混合溶液,滴至洁净载玻片上,置光学显微镜下,观察溶剂挥发过程中体系的动态变化,并录制视频,结果见图2。

图2 PC、EC、PE21 挥发过程图(20×)
由图2可知,0 min时三者均有细小微粒浮动;PC在溶剂挥发过程中微粒沉积并逐渐聚集成片,形成药物富集区域,待溶剂挥干后PC聚集区更密集。溶剂挥发过程中EC无明显聚集,与0 min相比有明显的细小颗粒分散分布,2 min25 s时溶剂挥干,可看出溶剂挥干后有大量粒径约1 µm的细小颗粒析出,终点时颗粒大部分呈均匀分布,少部分堆积。PE21在0 min出现稀疏的细小颗粒,1 min时与EC 3 min时现象类似,有密集细小颗粒均匀分布;随着溶剂挥发,发生相分离现象,呈现颗粒状和界限分明的沟壑状,可能是在此过程中EC包裹PC形成颗粒,而未被包裹的PC则以另一种形式存在,出现药物富集区。溶剂挥干后大部分区域呈均匀分布。
2.5 载药量和包封率的测定
准确称取5 mg微粒样品,加70 %乙醇使其充分溶解并定容至10 ml,按2.1项下方法测定原花青素含量;按以下公式计算微粒的载药量和包封率,平行测定3次,结果见表1。
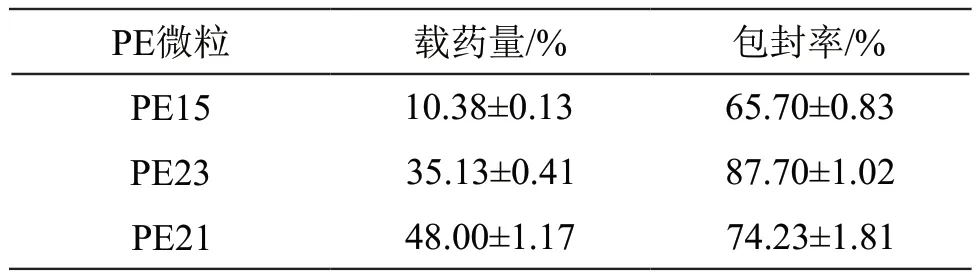
表1 PE微粒的载药量和包封率(n=3)
结果表明,PE微粒的包封率均较高,随着药物比例的增加,微粒载药量也在增加,但包封率先增后减。可能是因为EC具有黏性,使得载药微粒的载药量与包封率均较高。
2.6 傅立叶变换红外光谱(FT-IR)
采用105 ℃烘干5 h的溴化钾(KBr)混合研磨压片,扫描范围4000~400 cm-1,分辨率4.0 cm-1,扫描信号累加次数为16次;分别对PC、PC-MP、EC、EC-MP、PC与EC物理混合物(PE-PM)、PE15、PE23、PE21进行扫描,并绘制FT-IR谱图,结果见图3。

图3 FT-IR图谱
由图3可见,PC在3356 cm-1处为其分子结构中酚羟基由氢键作用引起的伸缩振动,1606,1519,1445 cm-1处是苯环骨架中C=C的伸缩振动吸收峰;1281,1152,1108,1060 cm-1对应的吸收峰为PC分子结构C环的C-O-C的伸缩振动;PC-MP与PC的红外吸收特征基本一致。EC在3477和2984 cm-1处的吸收峰由其分子中的-OH和-CH2伸缩振动产生,-CH3弯曲振动形成1380 cm-1处的峰。EC-MP相应位置的吸收峰均减弱。物理混合物的红外图谱基本为PC和EC的叠加。载药微粒的红外谱图基本一致且与原花青素谱图类似,表明载药微粒里的化合物主要以原花青素为主,苯环骨架C=C的伸缩振动吸收峰红移,载药微粒谱图在3416 cm-1处有吸收峰,这可能是因为原花青素与载体物质产生了分子间作用力,形成了氢键,-OH伸缩振动蓝移变为O-H伸缩振动产生的。
1606 cm-1处为PC的特征峰,在PE-PM图谱中的同一位置仍存在且强度增大,表明PC未被EC包裹并与其发生相互作用,与载药微粒相比,同一位置的峰仍存在但强度减弱,表明PC被EC包裹,且随PC的比例增加,1606 cm-1处的峰强度逐渐增大,表明有部分PC未被EC包裹,载药微粒可能会发生相分离现象。EC在3477,2984 cm-1处的吸收峰可为其特征峰,在PE-PM谱图中仍存在,但强度减弱,表明PC与EC物理混合时可能形成氢键,从而影响其强度。与载药微粒相比,同一位置的峰仍存在,但由于EC将PC包裹产生分子间作用力,使其强度更弱。
2.7 体外释放考察
分别精密称取一定量的PC-MP、PE21于5 ml离心管中,加入3 ml释放介质(pH 6.8磷酸盐缓冲液),离心管置于恒温培养摇床内(37±0.5 ℃,100 r/min),分别于0.5,1,2,4,6,8,10,12,24 h高速(10 000 r/min)离心5 min后,取上清(2.7 ml),同时补充同温等量介质,经0.22 µm水系针头式微孔滤膜过滤,取续滤液 1 ml,按2.1项下方法测定PC含量,平行测定3次,据PC标准曲线计算浓度,根据以下计算累积释放量(Q)[10],并以累积释放量对相应的时间作图,分别绘制体外释放曲线。
其中,V0为释放介质总体积;Vi为释放介质置换体积;Ci为在第i个时间点取样时释放介质中的药物浓度;m为微粒所载药物总质量;n为取样次数,结果见图4。

图4 原花青素缓释微粒累计释放曲线
由图4可知,两者在12 h时均已释放80 %,24 h释放较完全;PE21在0.5 h时释药略低于PCMP,此后均较高;PE21在12 h内的释放呈典型的缓释特征,而PC-MP在2 h内快速释放50 %之后,此后10 h内释药速率几乎恒定。
0.5 h之前PC-MP是直接释放,PE21因缓释材料EC的存在使得释放减慢,0.5 h之后PC-MP释药速率低于PE21。PE21的粒径为3~10 µm,PC-MP粒径10~25 µm,PE21粒径小、比表面积大,且EC具有黏性使载药微粒表面吸附有PC, 0.5 h后,微粒表面吸附及靠近微粒外层的药物迅速释放,使得PE21在0.5 h后的释放较快,此后,因EC的存在出现缓释特征。
2.8 模型拟合与释药机制分析
为了进一步阐明不同药载比原花青素微粒的体外释药特性,探讨其释药机理,在上述结果的基础上,采用4种动力学模型对PC-MP、PE21的释放曲线进行拟合[11],包括:零级动力学模型(Qt=kt)、一级动力学模型[ln(1-Qt)=-kt]、Higuchi模型(Qt=kt0.5)、Ritger-Peppas模型(Qt=ktn)。
式中:为t时刻的累积释放分数;k为释放常数;Ritger-Peppas模型中的n值表示释放机制,n≤0.45为菲克扩散(Fick diffusion),n≥0.89为骨架溶蚀,当n值介于二者之间为非菲克扩散(结合扩散与骨架溶蚀作用)[12]。根据所得方程的相关系数R2,判断释放方程拟合的最佳模型。
由表2可知,零级动力学模型拟合结果最差,表明药物与微粒释放不是恒定的即非控释;一级动力学模型拟合效果仅次于Ritger-Peppas模型,且PC-MP的R2大于PE21,原因可能是累计释放曲线PC-MP在2~12 h阶段为一级动力学释放。原花青素微粒的体外释放最佳模型为Ritger-Peppas模型,两者n值均小于0.45,因此其释放机制为菲克扩散,属于非稳态扩散。
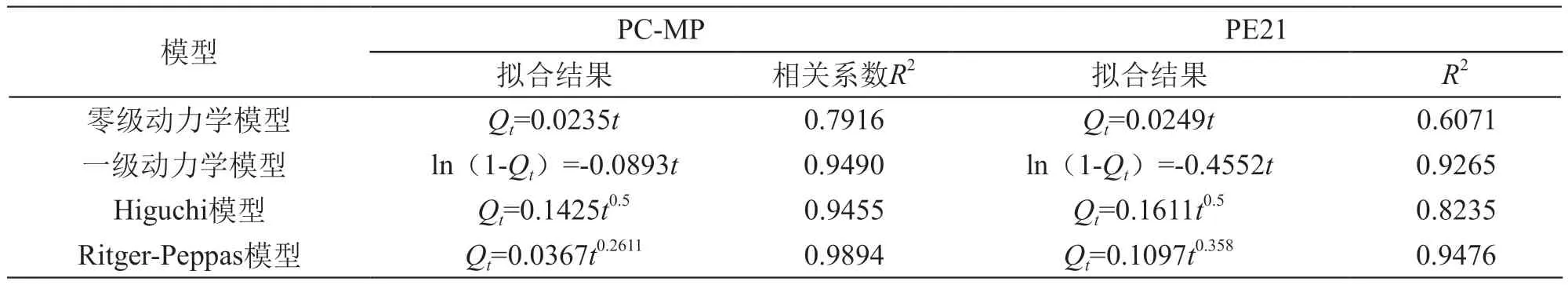
表2 原花青素缓释微粒体外释放的拟合结果
3 讨论
据图1结果推测生产PC时可能采用了喷雾干燥工艺,因而形成了大量中空球形粒子、且在快速固化过程中发生表面塌陷,这与文献报道中喷雾干燥过程中的条件、处方组成等多种因素造成可能造成微粒表面皱褶相符合[13-14]。而本文采用涂膜粉碎法制备微粒的过程中,将PC粒子复溶、自然挥干之后再粉碎,完全改变了PC原料药的原有形貌,与文献中采用涂膜粉碎法制备微胶囊的结果相似[9],均呈现不规则的颗粒且粒径较大。比较EC涂膜粉碎法处理前后,EC粒子的形态改变更明显,从酥松块状物转变为成片的细小均匀微粒,此结果与EC的特性一致。此法操作简单,可用于制备微米级且密度更小、力学性能更好的聚合物微粒。以上可表明不同的制备工艺对粒子形态有极大影响。
研究表明,当溶液中化合物浓度超过其无定形形式的溶解度时,就会在过饱和溶液中形成聚集体。在此浓度(通常为结晶溶解度的5~50倍)下,会超过化合物液体形式的混溶性极限,并发生液-液相分离(LLPS),从而形成初始为胶体大小的药物富集相[15]。因PC的极性比EC大,亲水性较EC好,在乙醇溶剂挥发剩余部分水时更易形成药物富集区;而EC不溶于水、极性小,在溶剂中乙醇挥发完之后产生相分离且比PC早,不易形成药物富集区;载药微粒因PC与EC发生相互作用形成分子间氢键, EC极性小于PC,且PC结构中有苯环不易变形,EC结构中无苯环易变形,在乙醇挥发之后EC不溶于剩余水分,使得EC包裹PC形成较规则且粒径较EC大的粒子,未被包裹的PC则形成药物富集区。还可能因EC具有黏性,可吸附PC形成药物富集区,在干燥过程中形成粘连结块。
本文采用涂膜粉碎法成功制备了载PC的EC缓释微粒,结果表明通过改变药载比能方便有效地调节所得载药微粒的形态,进而影响其载药和释药性能,证实涂膜粉碎用于制备载药微粒高效便捷、可控性强。本研究为涂膜粉碎工艺推广用于其他活性成分制剂开发奠定了基础,也对微粒结构和性能的调控策略进行了有益探索。
猜你喜欢
中华环境(2021年9期)2021-10-14
中华环境(2021年8期)2021-10-13
中华环境(2021年7期)2021-08-14
塑料包装(2021年3期)2021-01-25
疯狂英语·新悦读(2017年6期)2017-06-24
天然产物研究与开发(2016年6期)2016-06-05
化工进展(2015年3期)2015-11-11
华东理工大学学报(自然科学版)(2015年5期)2015-02-27
天然产物研究与开发(2014年6期)2014-04-27
食品工业科技(2014年21期)2014-03-11
