
HPLC指纹图谱结合化学计量学评价不同产地木棉花药材质量
2023-08-28 02:22黎桃敏陈丹燕余欣彤甘力帆张兰兰
食品与药品 2023年4期
黎桃敏,陈丹燕,余欣彤,甘力帆,张兰兰,黄 森,魏 梅*
(1. 广东一方制药有限公司,广东 佛山 528244;2. 广东省中药配方颗粒企业重点实验室,广东 佛山 528244)
木棉花又名攀枝花、红茉莉、红棉花、英雄树花,是木棉科植物木棉Gossampinus malabarica(DC.) Merr.的干燥花,性凉,具清热利湿、解毒功效,可用于治疗泄泻,痢疾,痔疮出血等症,主产于云南、广东、广西、海南、福建、台湾等地[1-2]。现代研究表明,木棉花药材的化学成分主要有黄酮类化合物、苯丙素类化合物、有机酸及酯类化合物等,在抗氧化、抗炎、抗肿瘤、保肝等方面有重要的药理作用[2-4]。
木棉花是我国南方地区常用的一味药食同源中药材,民间药用经验丰富[5-6],但由于其产地多、生长环境不同,药材质量难以控制。2020年版《中国药典》一部木棉花项下仅规定了性状、鉴别、浸出物检验项,难以判别药材质量优劣。现行的中药材质量控制模式主要有指标成分含量测定和指纹图谱技术[7-10]。目前已有木棉花花萼高效液相色谱(HPLC)含量测定方法的文献报道[11],但尚无采用HPLC指纹图谱评价木棉花药材质量的相关报道。本研究通过建立木棉花HPLC指纹图谱,结合化学计量学分析方法研究不同产地木棉花药材质量差异,明确质量差异标志物,以期为木棉花药材资源评价及质量控制提供依据。
1 仪器与试药
1.1 仪器
Agilent 1290型高效液相色谱仪(美国Agilent公司);XP26型百万分之一天平,ME204E型万分之一天平(瑞士Mettler Toledo公司);Milli-Q Direct型超纯水系统(德国Merck公司);KQ-500DE型数控超声波清洗器(昆山市超声仪器有限公司);111B型二两装高速中药粉碎机(浙江瑞安市永历制药机械有限公司)。
1.2 试药
原儿茶酸对照品(批号:110809-201906,纯度:97.7 %),芒果苷对照品(批号:111607-201704,纯度:98.1 %),芦丁对照品(批号:100080-201811,纯度:91.7 %,中国食品药品检定研究院);新绿原酸对照品(批号:151217,纯度:98.5 %,成都普菲德生物技术有限公司);乙腈,甲醇为色谱级(德国Merck公司),磷酸为色谱级(天津市科密欧化学试剂有限公司),水为超纯水,其余试剂均为分析纯。
15批木棉花药材(编号:Y1~Y15)经广东一方制药有限公司魏梅主任药师鉴定为正品,均符合2020年版《中国药典》一部木棉花项下要求。样品来源及编号分别为云南省(编号:Y1~Y6,Y10~Y12),海南省(编号:Y7~Y9),广西壮族自治区(编号:Y13~Y15)。
2 方法与结果
2.1 色谱条件[12]
色谱柱:Waters Xselect HSS T3柱(250 mm×4.6 mm,5 μm);流动相:乙腈(A)-0.5 %磷酸溶液(B),梯度洗脱(0~15 min,5 %A→10 %A;15~30 min,10 %A→17 %A;30~40 min,17 %A→18 %A;40~41 min,18 %A→5 %A;41~45 min,5 %A);流速:1.0 ml/min;柱温:25 ℃;检测波长:230 nm;进样量:10 μl。
2.2 对照品溶液的配制
分别取原儿茶酸、新绿原酸、芒果苷、芦丁对照品适量,精密称定,加入50 %甲醇,制成每1 ml分别含84.804,43.096,41.594,48.513 μg的混合对照品贮备液。另取芒果苷对照品适量,精密称定,加入50 %甲醇,制成每1 ml含芒果苷42.771 μg的对照品贮备液。
2.3 供试品溶液的制备
取木棉花药材粉末(过三号筛)约1.0 g,精密称定,置具塞锥形瓶中,精密加入50 %甲醇25 ml,称定重量,超声处理(功率250 W,频率40 kHz)45 min,放冷,再称定重量,用50 %甲醇补足减失的重量,摇匀,滤过,取续滤液,即得。
2.4 HPLC指纹图谱方法学考察
2.4.1 专属性试验 分别精密吸取空白溶剂、混合对照品溶液及供试品溶液,按2.1项下色谱条件进样测定,结果见图1。供试品溶液色谱在与对照品溶液色谱相应的保留时间处有相同的色谱峰,且空白溶剂无干扰,表明该方法专属性良好。
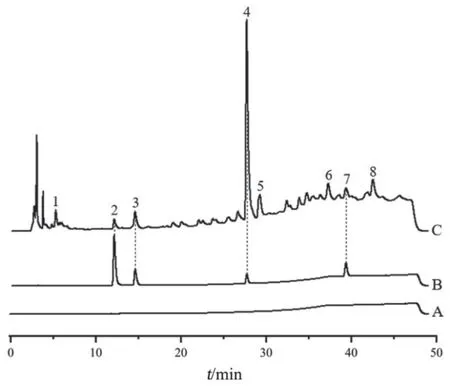
图1 专属性试验HPLC图谱
2.4.2 精密度试验 精密吸取2.2项下混合对照品溶液,按2.1项下色谱条件连续进样6次,记录色谱图。计算得各色谱峰峰面积的RSD均小于3.0 %,表明仪器精密度良好。
2.4.3 重复性试验 取木棉花药材(Y5)粉末,精密称定,平行称定6份,按2.3项下方法制备供试品溶液,按2.1项下色谱条件进样测定,记录色谱图。以4号芒果苷色谱峰为参照峰(S),计算得各共有峰相对保留时间和相对峰面积的RSD均小于3.0 %,表明该方法重复性良好。
2.4.4 稳定性试验 取木棉花药材(Y5)粉末,精密称定,按2.3项下方法制备供试品溶液,按2.1项下色谱条件,分别在0,2,4,6,8,12,24 h进样测定,记录色谱图。以4号芒果苷色谱峰为参照峰(S),计算得各共有峰相对保留时间和相对峰面积的RSD均小于3.0 %,表明供试品溶液在24 h内稳定性良好。
2.5 HPLC指纹图谱的建立及相似度评价
取15批木棉花药材,按2.3项下方法制备供试品溶液,按2.1项下色谱条件进样测定,记录色谱图。将15批木棉花药材色谱图导入《中药色谱指纹图谱相似度评价系统》(2012版)软件中,以Y1样品的色谱图为参照图谱,将时间窗宽度设为0.1 min,采用多点校正法进行色谱峰匹配,选择中位数法生成对照指纹图谱R,计算各批次木棉花药材与对照指纹图谱R的相似度。15批木棉花药材的HPLC指纹图谱见图2,相似度评价结果见表1。15批木棉花药材指纹图谱共标记了8个共有峰,通过与对照品图谱对比,指认了4个共有峰,分别为原儿茶酸(峰2),新绿原酸(峰3),芒果苷(峰4)和芦丁(峰7)。15批木棉花药材指纹图谱与对照指纹图谱R的相似度均大于0.990,表明不同批次木棉花药材的整体质量较一致,为进一步探究不同产地样品内在成分的差异性,还需结合化学计量学方法进行分析。
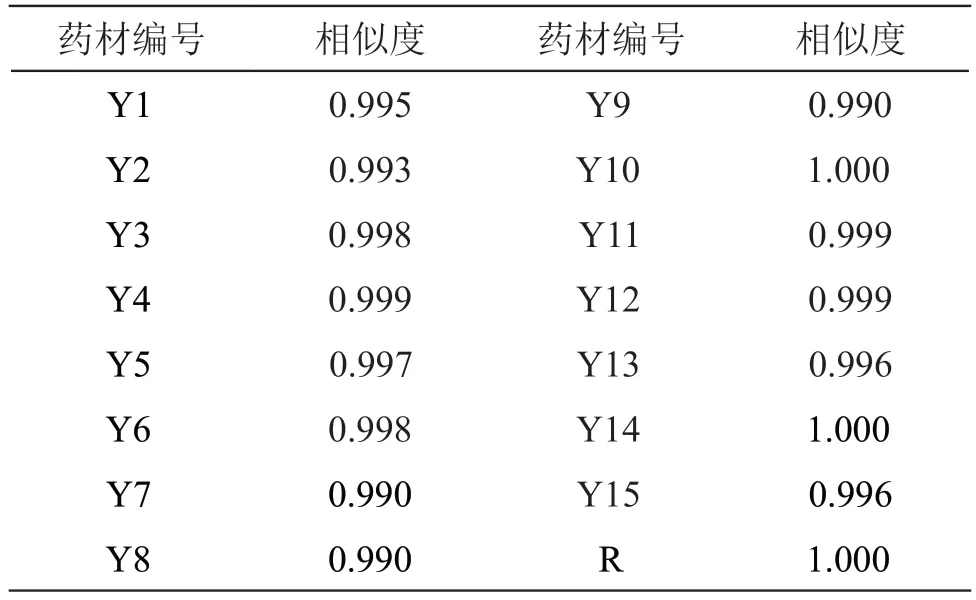
表1 15批木棉花药材HPLC指纹图谱的相似度评价结果
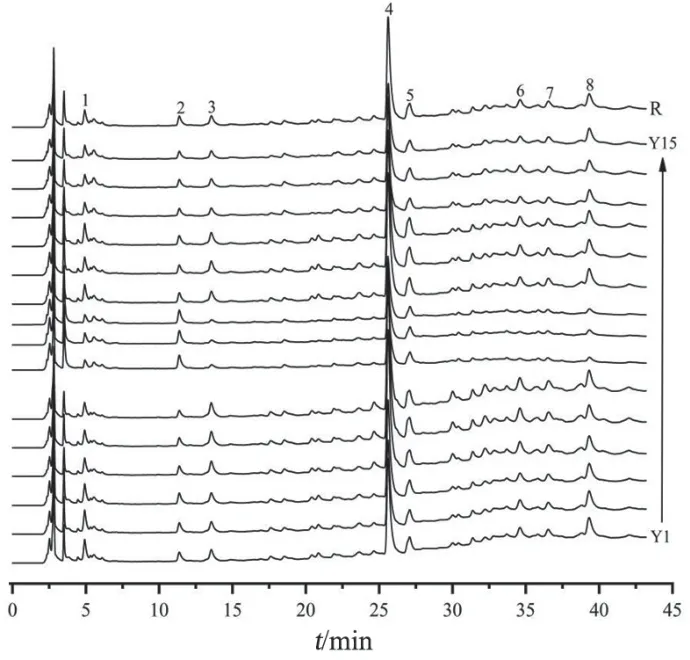
图2 15批木棉花药材的HPLC指纹图谱叠加图
2.6 化学计量学方法评价
2.6.1 热图聚类分析(CA) 以8个共有峰的峰面积为变量,经标准化处理后,采用线性归一化法,以Euclidean平方距离为度量标准,利用HemI 1.0软件对15批木棉花药材指纹图谱进行热图和聚类分析,结果见图3。
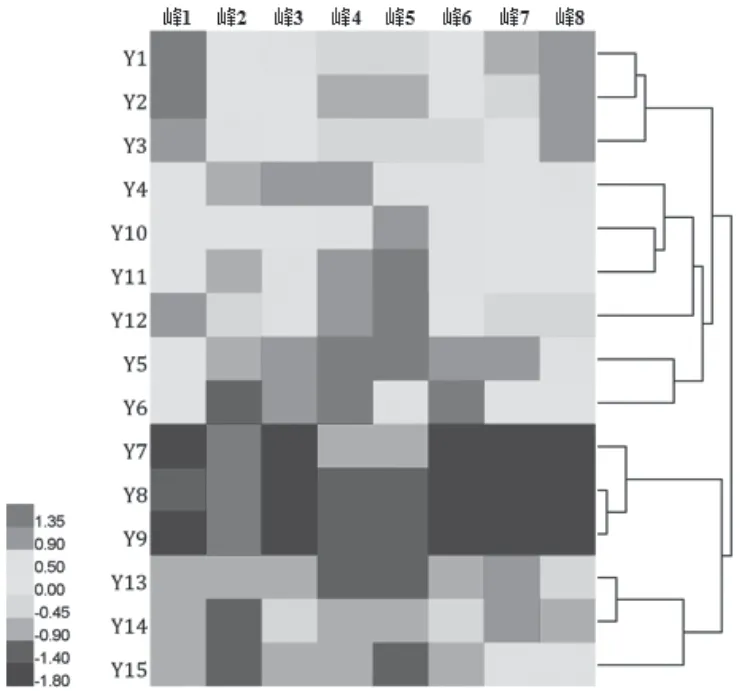
图3 15批木棉花药材HPLC指纹图谱热图聚类分析图
聚类分析结果显示15批木棉花药材可大致聚为2类,其中海南3批药材(Y7~Y9)聚为I-a类,广西3批药材(Y13~Y15)聚为I-b类,9批云南药材(Y1~Y6、Y10~12)聚为II类。说明不同产地的木棉花药材之间存在一定差异,海南与广西的药材质量更接近。
热图分析以一种渐进的蓝红色将结果直观展现出来,其颜色代表归一化后的值,值越高颜色越红,值越低颜色越蓝,可用于反映研究对象化学成分的含量高低[13-14]。热图分析结果显示不同产地木棉花药材的化学成分含量差异较大,其中云南9份样品(Y1~Y6、Y10~12)各特征峰颜色整体偏暖,表明其各成分含量普遍较高;广西3份样品(Y13~Y15)各特征峰颜色整体偏冷,表明其各成分含量普遍较低;海南3份样品(Y7~Y9)的峰2(原儿茶酸)颜色偏暖,其他特征峰颜色整体呈蓝色,说明除原儿茶酸外,其他成分的含量普遍较云南、广西要低。
2.6.2 主成分分析(PCA) 主成分分析可根据实际需要提取少数几个互相独立的综合指标变量,以尽可能反映原有变量信息,是一种科学有效的降维方法[15]。以各共有峰的峰面积为变量,采用SPSS 20.0软件对15批木棉花药材的峰面积数据作标准化处理,并进行因子分析,计算得特征值和方差贡献率,见表2,旋转成分矩阵见表3。以特征值>1为提取标准提取主成分,提取出前2个主成分,主成分1和主成分2的累积方差贡献率达83.938 %,表明前2个主成分能反映样品中绝大部分的原变量信息,是影响木棉花药材质量差异的重要因素。由表3可知,以载荷系数绝对值≥0.60为选取标准,主成分因子1所反映的色谱峰有峰1、新绿原酸、芒果苷、峰5、峰6和峰8,主成分因子2所反映的色谱峰有原儿茶酸和芦丁。
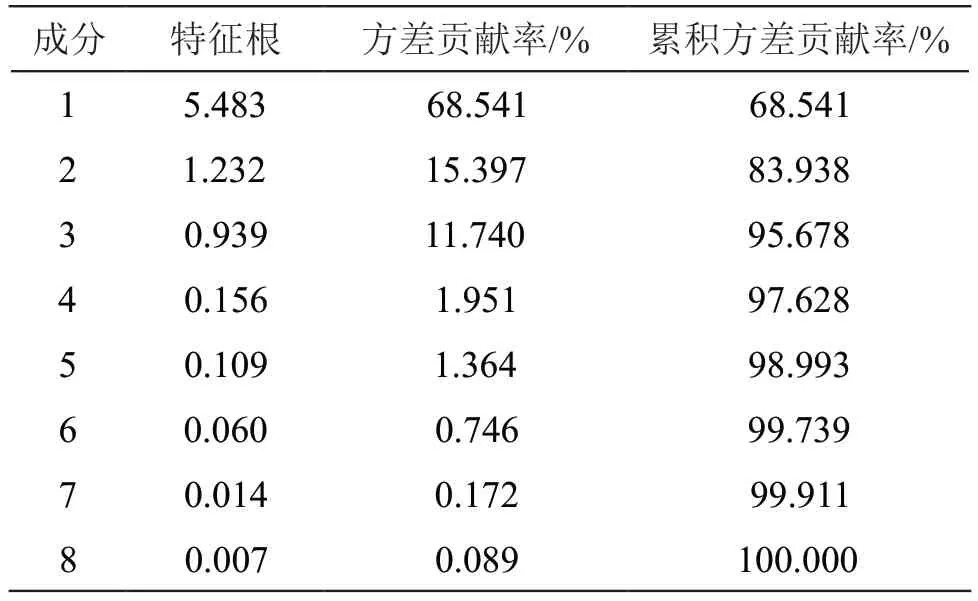
表2 特征值和方差贡献率
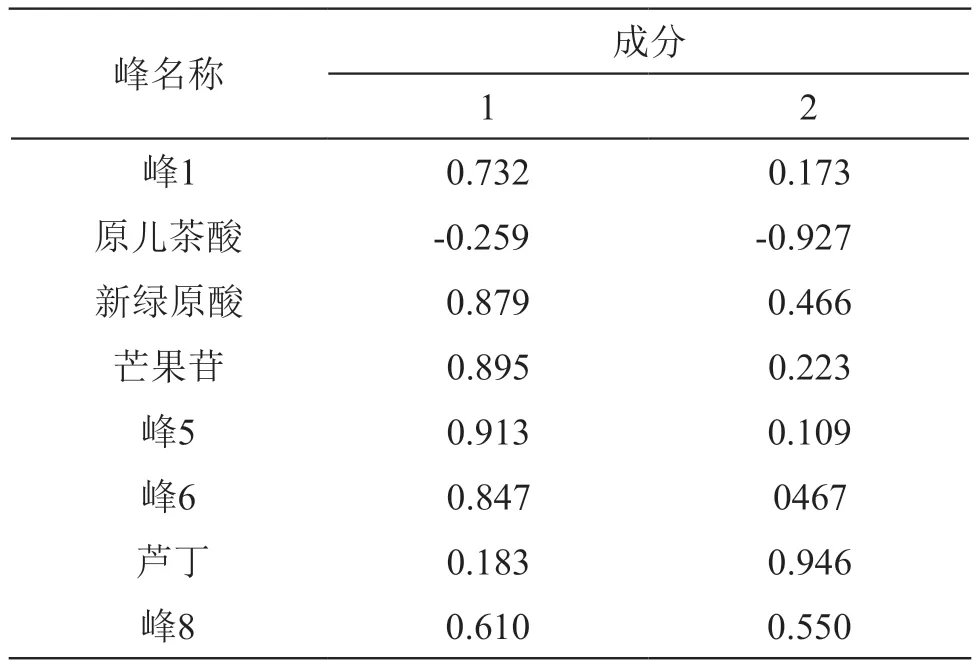
表3 旋转成分矩阵
2.6.3 正交偏最小二乘法判别分析(OPLS-DA)为进一步筛选影响不同产地木棉花药材质量差异的标志物,将15批木棉花药材的共有峰峰面积导入SIMCA 14.1软件,采用监督模式识别法进行OPLSDA,并绘制OPLS-DA模型得分图,见图4。所得OPLS-DA模型中的拟合参数R2X=0.956,R2Y=0.952,模型预测参数(Q2)=0.927>0.5,表明建立的模型良好且预测能力较强。样品分布距离越远表明差异性越大[16],由图4可见,3个产地药材间的距离均较远,各自聚为一类,表明不同产地的木棉花药材之间存在明显差异。
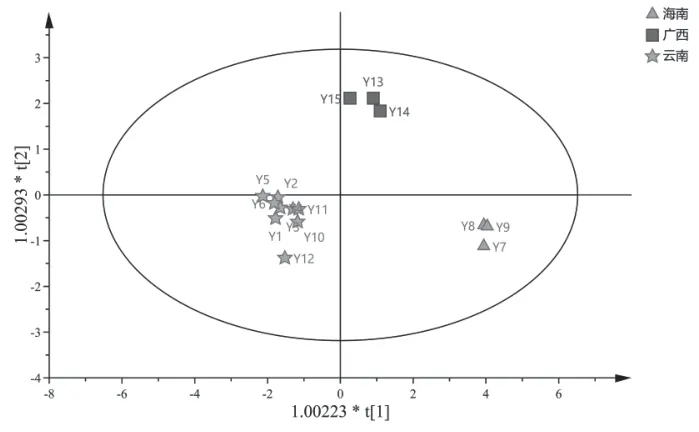
图4 15批木棉花药材HPLC指纹图谱的OPLS-DA得分图
以模型变量投影重要性(VIP)值>1为指标筛选15批木棉花药材的差异成分,见图5。VIP值越高,对组间差异的影响越大。结果显示峰4和峰8的VIP值大于1,说明这2个色谱峰所代表的化学成分是引起不同产地木棉花药材质量差异的标志物。由于2个色谱峰只指认了4号色谱峰为芒果苷,故选择芒果苷作为质量差异成分对15批木棉花药材进行含量测定。
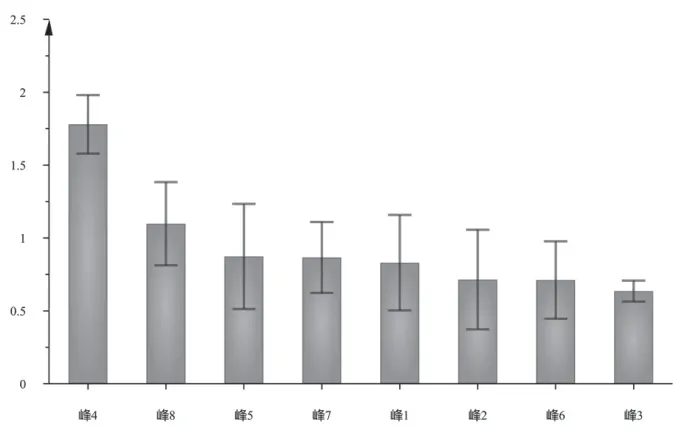
图5 15批木棉花药材HPLC指纹图谱的OPLS-DA VIP值
2.7 芒果苷含量测定
2.7.1 线性关系考察 取芒果苷对照品适量,精密称定,加入50 %甲醇,制成每1 ml含芒果苷285.275 μg的对照品母液。精密吸取芒果苷对照品母液适量,加50 %甲醇制成系列质量浓度的对照品溶液,按2.1项下色谱条件进样测定,记录色谱图,以对照品溶液质量浓度(μg/ml)为横坐标(X),峰面积为纵坐标(Y),绘制标准曲线,得芒果苷线性回归方程为Y=2.158×103X-2.793×103(r=0.9993),结果表明芒果苷质量浓度在2.853~285.275 μg/ml范围内线性关系良好。
2.7.2 精密度试验 精密吸取2.2项下芒果苷对照品溶液,按2.1项下色谱条件连续进样6次,记录色谱图。计算得芒果苷峰面积的RSD为0.45 %,表明仪器精密度良好。
2.7.3 重复性试验 取木棉花药材(Y5)粉末,精密称定,平行称定6份,按2.3项下方法制备供试品溶液,按2.1项下色谱条件进样测定,记录色谱图。计算得芒果苷含量的RSD为0.51 %,表明该方法重复性良好。
2.7.4 稳定性试验 取木棉花药材(Y5)粉末,精密称定,按2.3项下方法制备供试品溶液,按2.1项下色谱条件,分别在0,2,4,6,8,12,24 h进样测定,记录色谱图。计算得芒果苷含量的RSD为0.32 %,表明供试品溶液在24 h内稳定性良好。
2.7.5 加样回收试验 取已知芒果苷含有量的木棉花药材粉末,精密称取9份,每份约0.5 g,置具塞锥形瓶中,均分为3组,每组分别精密加入相当于含有量50 %,100 %,150 %的芒果苷对照品溶液,按2.3项下方法制备供试品溶液,按2.1项下色谱条件进样测定,计算加样回收率及RSD,结果见表4,芒果苷的平均加样回收率为101.04 %,RSD为1.57 %,表明该方法准确度良好。
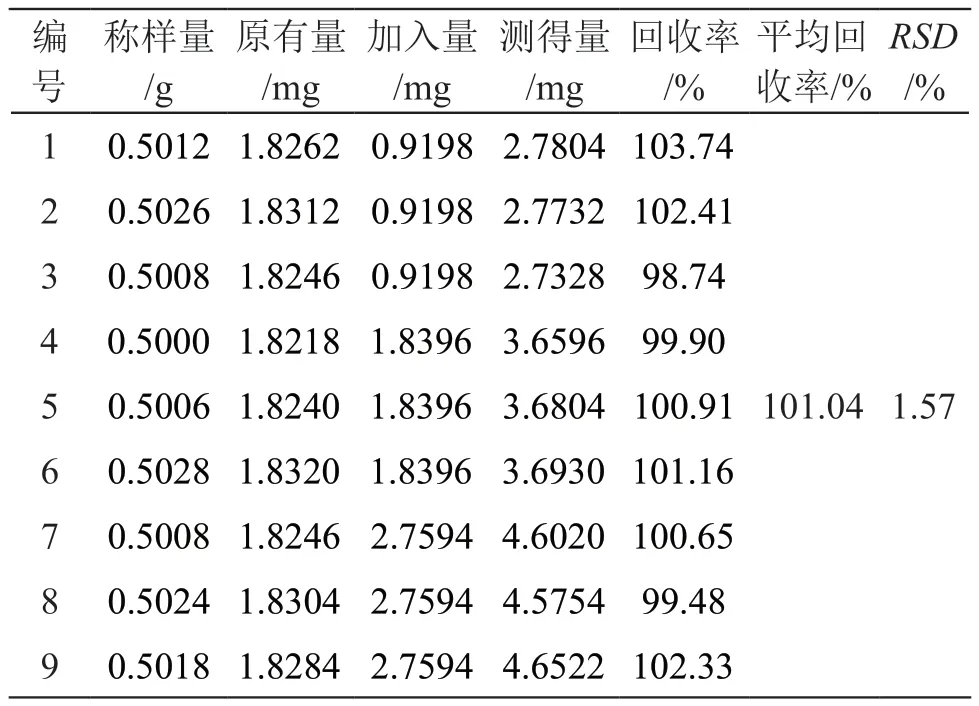
表4 芒果苷加样回收试验结果(n=9)
2.8 Spearman相关分析
取15批木棉花药材,按2.3项下方法制备供试品溶液,按2.1项下色谱条件进样测定,芒果苷含量测定结果见表5。基于PCA模型,将主成分因子得分与其方差贡献率乘积之和相加[17],计算15批木棉花药材指纹图谱的综合评分F,结果见表5。采用SPSS 20.0软件对15批木棉花药材的芒果苷含量与其指纹图谱综合评分进行Spearman相关分析,结果两者的相关系数r为0.975(P<0.01),表明芒果苷含量与指纹图谱综合评分呈显著正相关性。由表5可见,芒果苷含量和指纹图谱综合得分排名前9的木棉花药材均为云南产地,表明云南产地的木棉花药材质量较好。
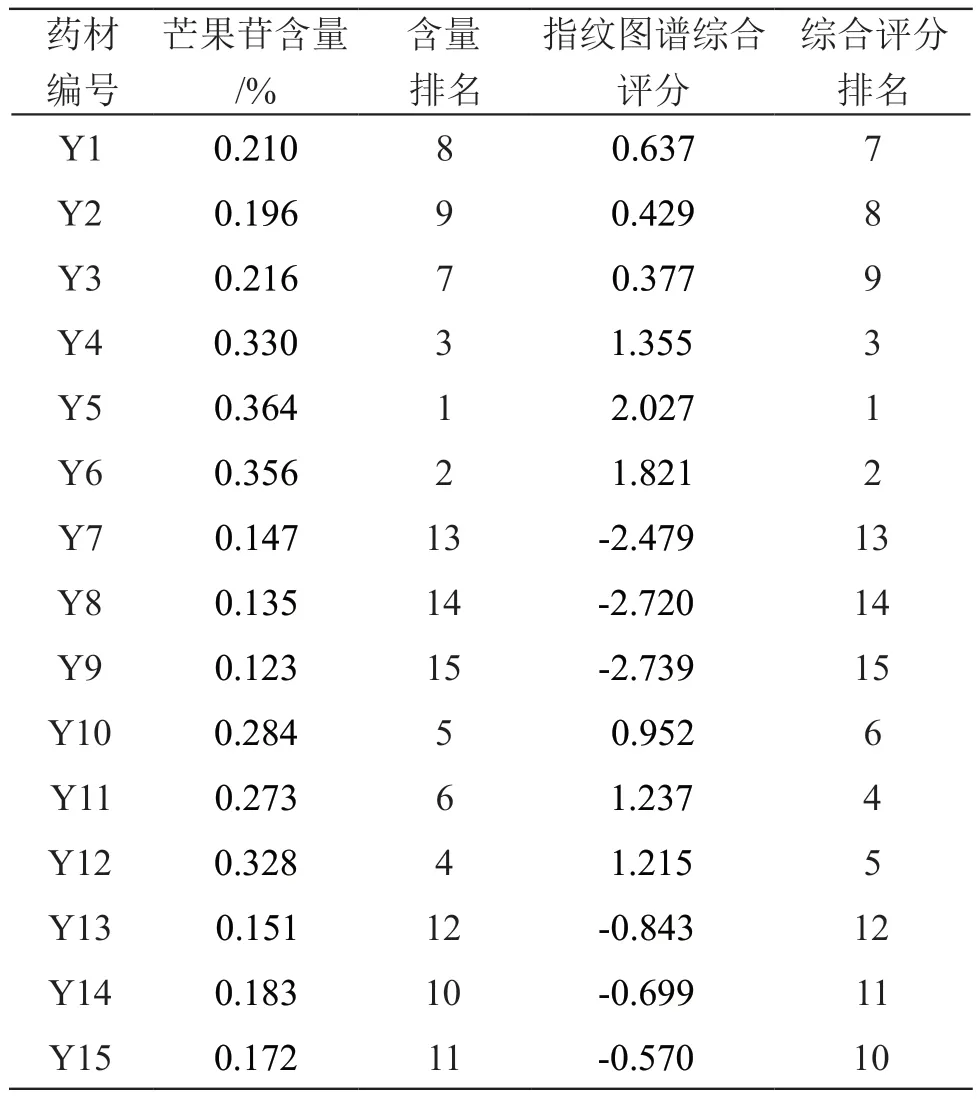
表5 15批木棉花药材芒果苷含量及指纹图谱综合评分结果
3 讨论
研究结果表明,15批木棉花药材指纹图谱的CA结果分为2类,海南样品与广西样品分别聚为I-a、I-b类,云南产样品聚为II类。说明3个产地的药材质量差异较大,其中海南与广西的药材质量相对较接近,这可能与地理位置、气候环境等因素有关。热图分析结果显示不同产地间木棉花药材的化学成分含量存在差异。经OPLS-DA筛选出2个主要影响药材质量的差异成分,通过对照品的指认,仅可确定其中1种成分(芒果苷),后续可结合液质联用技术,对未知的色谱峰成分进行指认。指纹图谱综合评分、芒果苷含量测定等结果均表明不同产地木棉花药材质量存在一定差异,其中云南产的木棉花药材质量更优。另本研究收集到的木棉花药材样品共15批,所属产地仅3个,后续可增加多个其他产地的样品,并加大样本量进行统计分析,以获得可靠的数据,为筛选优良种质木棉花药材提供参考。
近年,化学计量学在中药制剂各环节和质量评价中的应用愈为常见,其中在药材收集环节中的应用,旨在对药材进行分类和鉴别,从药材源头控制中药产品的质量,以保证用于制备中药制剂的原药材一致性良好。本研究构建的指纹图谱结合化学计量学方法的综合评价体系,能成功应用于不同产地木棉花药材的质量评价,可为木棉花药材资源评价提供理论支撑,也对木棉花相关制剂生产过程的质量控制具有一定的指导作用。
猜你喜欢
人民之声(2022年3期)2022-04-12
歌海(2021年2期)2021-06-22
动漫界·幼教365(中班)(2020年3期)2020-04-20
中国外汇(2019年10期)2019-08-27
中国外汇(2019年10期)2019-08-27
中国外汇(2019年22期)2019-05-21
中国外汇(2019年21期)2019-05-21
课堂内外·创新作文小学版(2018年7期)2018-08-16
动漫界·幼教365(小班)(2018年10期)2018-05-14
动漫界·幼教365(大班)(2018年10期)2018-05-14
