
miR-361-3p调控BTG2对HL-60细胞增殖和凋亡的影响*
2022-12-22 03:16许汇娟李珍
广东医学 2022年11期
许汇娟, 李珍
1南方医科大学南方医院血液科(广东广州 510515); 2南方医科大学检验与生物技术学院(广东广州 510515)
急性髓细胞白血病(acute myeloid leukemia,AML)是髓系造血干/祖细胞恶性疾病,通常以骨髓和外周血中原始和(或)幼稚髓性细胞异常增生为主要特征,大多数病例病情急重,预后凶险,如不及时治疗常可危及生命[1]。因此,寻找白血病治疗靶点,开发有特异性的靶向药物具有重要的临床意义。微小RNA (microRNA,miRNA)是一类大约22个碱基长的非编码RNA。miRNA通过特异性抑制或降解靶基因的翻译,调节靶基因的表达[2]。越来越多的证据表明,miRNA参与人体内生理和病理的过程,比如miRNA在细胞分化、新陈代谢以及肿瘤的发生和发展中发挥重要作用[3-4]。在人类癌症中,miRNA的功能可根据靶基因的不同,作为调控肿瘤发生发展的癌基因或抑癌基因[5]。miR-361被认为是多种肿瘤的致癌因子。研究[6]报道,通过比较14种肿瘤共5727例患者样本与589例正常人样本,发现miR-361在9种肿瘤组织中有高表达。另有研究通过miRNA/mRNA网络分析表明miR-361与白血病存在密切相关性[7],并且miR-361在急性白血病患者骨髓中也有显著上调[8]。尽管有很多研究提示miR-361是一种肿瘤因子,在多种恶性肿瘤中有高表达,并且在白血病患者体内表达上调,与白血病密切相关,但是,miR-361-3p 对急性髓系白血病细胞系HL-60细胞增殖的影响,及其靶向分子机制尚鲜见报道。2018年4月至2021年4月,本研究采用了荧光素酶报告基因检测等技术验证miR-361-3p和靶基因之间的关系,为阐明miR-361-3p调节白血病细胞HL-60增殖和凋亡的分子机制提供依据。
1 材料与方法
1.1 材料 HEK-293和HL-60细胞购自中国科学院上海细胞库,T4连接酶、限制性内切酶 XhoⅠ和NotⅠ购自Thermo公司。双荧光素酶报告基因载体(pmirGLO载体)和LipofectamineTM2000购自美国Invitrogen公司。大肠杆菌DH5α,Phanta®Max Super-Fidelity DNA Polymerase购自南京诺唯赞生物科技有限公司。DL2000bp Marker,DMEM培养基,Gel Extraction胶回收试剂盒,胎牛血清和Plasmid Mini KitI 质粒提取试剂盒购自美国OMEGA公司,miR-361-3p和PCR引物购自上海生工生物技术有限公司,双荧光素酶报告基因荧光检测试剂盒购自美国Promega公司。CCK-8和细胞凋亡检测试剂盒购自日本Dojindo公司,GAPDH、二抗:caspase-3、caspase-7、P53、Bcl-2、survive 等抗体均购自美国abcam公司。
1.2 方法
1.2.1 细胞培养 HEK-293和HL-60细胞用含10%胎牛血清的DMEM培养基培养,加入1%双抗(100 U/mL青霉素、100 U/mL链霉素),置于37℃,5%CO2培养基中培养。
1.2.2 靶基因预测 通过靶基因预测数据库starbase(http://starbase.sysu.edu.cn)预测miR-361-3p的靶基因,综合文献分析选定与细胞增殖凋亡相关的基因,包括B细胞迁移基因2抗体(B-cell translocation gene 2,BTG2),胞浆多聚元件结合蛋白1(cytoplasmic polyadenylation element binding protein 1,CPEB1)和髓磷脂转录因子1(myelin transcription factor,MYT1)作为miR-361-3p的候选靶基因。
1.2.3 miR-361-3p靶基因荧光报告载体的构建 设计合成BTG2, CPEB1 和MYT1上下游特异性同源引物,如表1。以HL-60细胞cDNA为模板,分别用 BTG2、CPEB1 和MYT1上下游特异性引物扩增BTG2, CPEB1 和MYT1的 3′UTR序列。分别取BTG2, CPEB1 和MYT1基因Polymerase Chain Reaction(PCR)回收产物和psiCHECK-2载体,用XhoⅠ与NotⅠ进行双酶切。酶切产物经2%琼脂糖凝胶电泳检测后,回收纯化。按照Plasmid Mini Kit Ⅰ质粒提取试剂盒说明书进行目的片段与载体连接,于大肠杆菌DH5α感受态细胞中加入10 μL连接产物,转化、挑取单菌落扩增,再进行双酶切、鉴定和测序(上海生物生工有限公司广州分公司)。

表1 BTG2、CPEB1 和MYT1引物序列
1.2.4 HEK-293细胞培养及转染 将HEK-293细胞接种在24孔板中(8×105·mL-1),培养过夜,用于共转染试验。将细胞随机分组为:野生型+miR-361-3p mimic组、野生型+空质粒组、突变型+miR-361-3p mimic、突变型+空质粒组,每组分别设3个复孔。每孔用无血清培养基稀释,加入 lipofectamine 2000 2 μL,总体积为125 μL;另取每孔各加入8 μg不同质粒,加入DMEM培养基至总体积125 μL;然后将两种稀释液混合,室温下静置20 min;每孔加入250 μL转染复合液,混匀;在 5% CO2,37℃培养箱中培养5 h,去除含有转染复合物的培养基后,加入完全培养基继续培养48 h。
1.2.5 双荧光素酶报告基因活性检测 HEK-293细胞转染48 h后,去除培养基,用PBS(phosphate buffer saline)清洗2次,每孔加入100 μL的PBL(Passive Lysis Buffer),振摇,收集细胞裂解液。在发光板中每孔加入20 μL细胞裂解液,100 μL LARⅡ工作液,混匀,检测荧光素酶活性值;每孔再加入100 μL终止液,检测荧光素酶活性值,计算前后的比值。
1.2.6 细胞增殖和凋亡检测 使用Lipofectamine 2000转染试剂盒进行转染,在HL-60细胞中分别转染miR-361-3p mimics/mimic-NC、miR-361-3pinhibitor/inhibitor-NC或si-BTG2/si-NC后,培养24、48和72 h。然后加入CCK-8(Cell Counting Kit-8),37℃孵育1 h,用酶标仪测定波长450 nm下的吸光度。48 h后,采用流式细胞仪检测HL-60细胞的凋亡情况。
1.2.7 实时定量PCR检测miR-361-3p和BTG2基因表达量 用Trizol法提取各组HL-60细胞中的总RNA,然后根据逆转录试剂盒的使用说明书获得cDNA。取cDNA和引物,按照实时荧光定量PCR说明书配制反应体系。U6为miR-361-3p的内参,GAPDH为BTG2的内参。采用2-ΔΔCt法计算其相对表达。反应条件为95℃预变性30 s,40个循环扩增反应(94℃ 30 s,65℃ 30 s,94℃ 30 s),miR-361-3p引物序列为5′-CTCAACTGGTGTCGTGGAGTCGGCAATTCAGTTGAGAAATCAGA-3′(正向)、5′-ACACTCCAGCTGGGTCCCCCAGGTGTGATTCTG-3′(反向),BTG2引物序列为5′-ACGGGAAGGGAACCGACAT-3′(正向)、5′-CAGTGGTGTTTGTAGTGCTCTG-3′(反向),U6引物序列为5′-CTCGCTTCGGCAGCACA-3′(正向)、5′-AACGCTTCACGAATTTGCGT-3′(反向),GAPDH引物序列为5′-TGTTCGTCATGGGTGTGAAC-3′(正向)、5′-ATGGCATGGACTGTGGTCAT-3′(反向)。
1.2.8 免疫印迹(Western blotting,WB)检测蛋白表达 收集各组HL-60细胞进行裂解,提取蛋白,将蛋白溶液和缓冲溶液按 4∶1 的比例混合,煮沸10 min,等到细胞总蛋白样品。随后进行上样、电泳和转膜,加脱脂奶粉封闭1 h。加入一抗,4℃条件下过夜,TTBS漂洗3次×10 min,加入二抗,室温封闭 2 h。取出 PVDF 膜,TTBS漂洗,DAB 显色后照相。
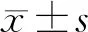
2 结果
2.1 重组载体质粒的构建 靶基因以HL-60细胞DNA为模板进行扩增,在200~250 bp左右有 DNA 条带,与预期的片段大小一致,见图1-A、1-C和1-E。将双酶切后的重组质粒进行琼脂糖凝胶电泳,结果如图1-B、1-D和1-F所示,证实BTG2、CPEB1和MYT1基因3′UTR已扩增出来。双酶切后野生型质粒和突变型质粒片段大小一致、分子量一致,说明两种载体构建成功。将筛选出的阳性克隆测序,测序结果进行BLAST比对,结果100%匹配。基因BTG2、CPEB1和MYT1已成功在目的突变区域实现突变,BTG2、CPEB1和MYT1基因3′UTR突变型部分测序图如图1-G、H。

注:A:BTG2基因 PCR产物电泳图;B:BTG2基因双酶切电泳图;C:CPEB1基因 PCR产物电泳图;D:CPEB1基因双酶切电泳图; E:MYT1基因 PCR产物电泳图;F:MYT1基因双酶切电泳图; G:BTG2基因3′UTR突变型部分测序图;H:CPEB1基因3′UTR突变型部分测序图;I:MYT1基因3′UTR突变型部分测序图
2.2 双荧光素报告基因系统分析miR-361-3p与靶基因的作用关系 通过Starbase检索发现,BTG2、CPEB1和MYT1 3′UTR与miR-361-3p存在结合位点。如表2所示,与 NC对照组相比,共转染突变型BTG2 3′UTR载体和miR-361-3p mimic组的荧光素酶活性变化差异无统计学意义(P>0.05);与此相反,共转染野生型BTG2 3′UTR载体和miR-361-3p mimic组的荧光素酶活性显著降低(P<0.001)。另外,与NC对照组相比,共转染突变型CPEB1和MYT1 3′UTR载体和miR-361-3p mimic组的荧光素酶活性差异无统计学意义(P>0.05);共转染野生型CPEB1和MYT1 3′UTR载体和miR-361-3p mimic组的荧光素酶活性降低,但差异无统计学意义(P>0.05)。
表2 荧光酶活性
2.3 miR-361-3p/BTG2轴调控HL-60细胞增殖和凋亡 与NC对照组相比,在HL-60细胞中转染miR-361-3pmimic后,细胞中miR-361-3p的表达水平显著升高(P<0.01);在HL-60细胞中转染miR-361-3pinhibitor,细胞中miR-361-3p的表达水平显著降低(P<0.01),见表3。并且,在HL-60细胞中转染miR-361-3pmimic,细胞中BTG2的表达水平显著降低,反之,在HL-60细胞中转染miR-361-3pinhibitor,细胞中BTG2的表达水平显著升高(P<0.01),见表4。CCK-8实验结果表明,miR-361-3p mimics能促进细胞增殖,与NC对照组相比,24、48和72 h的细胞增殖明显(P<0.01);与此相反,miR-361-3p inhibitor可抑制细胞增殖。同时,沉默BTG2也能促进HL-60细胞增殖(表4)。流式细胞术结果显示(表4和图2),与NC对照组相比,miR-361-3p mimic组和si BTG2组HL-60细胞凋亡比值下降,而miR-361-3p inhibitor组,细胞凋亡比值显著升高了(P<0.01)。

图2 流式细胞术检测各组HL-60细胞中细胞凋亡图
表3 各组HL-60细胞中miR-361-3p表达水平

表4 各组HL-60细胞中BTG2 mRNA表达水平、细胞增殖和细胞凋亡值
2.4 miR-361-3p/BTG2轴调控P53信号通路 与NC对照组相比,沉默BTG2后,HL-60细胞中P53、促凋亡因子caspase3/7蛋白表达水平均显著下调,差异有统计学意义(P<0.05)。反之,凋亡抑制因子Bcl-2、survive的蛋白表达水平显著上调,差异有统计学意义(P<0.05)。见图3和表5。
表5 各组HL-60细胞中caspase-3、caspase-7、P53、Bcl-2和survivine蛋白表达水平

图3 Western blot 检测HL-60细胞中caspase 3、caspase 7、p53、Bcl2和survivine蛋白的表达
3 讨论
MiRNA是指一大类负调控基因表达的非编码蛋白质的小RNA。它们在许多生物功能中起着至关重要的作用,如细胞生长、增殖、分化和凋亡[2-3]。人类多种恶性肿瘤(包括AML)的发生和发展与miRNAs的表达异常密切相关。一个miRNA能够靶向调控多个靶基因,从而发挥多种生物学功能。miR-361-3p在多种恶行肿瘤中表达显著性上调,起着癌基因的作用。2013年,Du等[9]首次报道了miR-361-3p是一种新的促癌基因。之后,相继有研究表明,miR-361-3p可调节人肺肿瘤细胞、前列腺癌细胞和口腔鳞状细胞癌(OSCC)的增殖和迁移[10]。例如,miR-361-3p通过负调控双特异性磷酸酶2(DUSP2)促进EMT,激活胰腺癌细胞中的ERK信号,从而促进细胞增殖[11]。这项研究还提供了临床证据,表明高水平的miR-361-3p与肿瘤晚期和较短的存活率有关。miR-361在新诊断的 AML 患者的血浆中显著增加(与健康对照组相比),经化疗后显著降低[12]。因此,我们推测miR-361-3p可能是一种AML 潜在的治疗靶点,它的靶向分子机制尚需进一步研究。为了探究miR-361-3p的靶基因,本研究通过生物信息学软件预测和相关文献分析,选取增殖凋亡相关的3个基因。进一步通过靶基因的3′UTR克隆至双荧光酶报告基因载体上,然后将构建成功的报告基因与miR-361-3p mimics 共转染HEK-293细胞中,双荧光素酶活性检测结果显示,miR-361-3p与BTG2有直接的靶向作用关系。
BTG2是BTG/TOB基因家族中第一个识别的基因,属于抗增殖基因家族[13]。BTG2通过几个重要的信号通路调节细胞生长、死亡、迁移、凋亡。同时,大量文献报道了在多种不同类型的癌细胞中存在BTG2 基因表达水平降低甚至缺失,与之相反,在其相应的正常细胞中 BTG2 基因表达水平明显较高,由此推测BTG2基因的表达异常与肿瘤具有密切关系,这提示了肿瘤的形成可能与 BTG2 基因表达相关[13-14]。在淋巴血液恶性肿瘤中,也发现存在BTG1/BTG2基因的缺失[15]。多项研究报道了,在HL-60细胞中,BTG2的高水平表达与抑制细胞增殖,促进细胞凋亡和分化相关[16-17]。为了探究miR-361-3p对白血病细胞增长的影响是否由BTG2介导,我们用miR-361-3p mimics/inhibitor或si-BTG2转染HL-60细胞后进行细胞增殖或凋亡的检测,结果证实了miR-361-3p/BTG2轴可调控白血病细胞的增殖和凋亡。
P53通路是重要的抑癌信号通路,它可通过调节细胞周期进程和(或)细胞凋亡等发挥抑癌作用[18]。我们进一步在蛋白水平验证沉默BTG2是否影响P53及其下游相关因子的表达。研究结果提示了miR-361-3p/BTG2轴在白血病HL-60细胞中可调控P53信号通路。综上所述,miR-361-3p通过BTG2调控P53通路影响白血病细胞的增殖和凋亡。这进一步为miR-361-3p/BTG2作为潜在的靶点被用于临床AML的诊断和治疗,提供更多的研究资料和实验依据。
利益相关声明:论文的全部作者声明,在课题研究和论文撰写过程中不存在利益冲突。
作者贡献说明:实验设计及论文撰写由李珍完成;实验实施,数据分析统计及论文撰写由许汇娟完成。
猜你喜欢
天津医科大学学报(2021年4期)2021-08-21
中日友好医院学报(2021年1期)2021-04-14
山东医药(2020年9期)2020-05-20
广西医学(2020年13期)2020-03-04
中国病理生理杂志(2018年9期)2018-09-27
河南畜牧兽医(2017年12期)2017-11-13
中国科技信息(2015年6期)2015-11-10
中国现代医生(2015年5期)2015-03-31
中国医药导报(2015年27期)2015-02-28
高中生学习·高三版(2014年3期)2014-04-29
