
负载型LaCoO3/MO2 催化氧化甲苯与NO 的性能研究
2022-08-04 14:53张俊丰赵令葵李思密陶泓帆伍云凡
燃料化学学报 2022年7期
张 娜 ,黄 妍 ,张俊丰 ,赵令葵 ,李思密 ,陶泓帆 ,伍云凡
(湘潭大学 环境与资源学院, 湖南 湘潭 411105)
挥发性有机化合物(VOCs)与氮氧化物(NOx)是引发光化学烟雾的重要前驱体,大量排放会严重威胁生态环境和人类健康[1,2]。随着交通运输业的飞速发展,柴油车排放的NOx和VOCs 对大气污染的贡献率逐渐增高,开发新型高效的柴油车排放控制技术势在必行。根据中国柴油车尾气处理技术规范,柴油车尾气后处理系统由催化氧化器(DOC) + 颗粒捕集器(DPF) + 选择性催化还原(SCR)组成,其中,DOC 装置主要是将尾气中的VOCs 和CO 催化氧化为CO2和H2O。通常,柴油车尾气中还含有一定量的NO,如能利用DOC将50% 以上的NO 转化为NO2,将有利于后续的DPF 再生和SCR 脱硝反应[3]。典型柴油车尾气组成的相关研究表明,甲苯是柴油车尾气排放VOCs 的代表性物质,占总量的30%−62%[4],因此,研究DOC 催化氧化VOCs 性能时,常以甲苯为模型污染物。
现有商用DOC 催化剂以铂(Pt)或钯(Pb)等贵金属为活性组分,催化氧化活性较高,但仍存在热稳定性差、成本高且易中毒等问题,严重制约着其应用[5],非贵金属DOC 催化剂研发备受关注[6,7]。钙钛矿型氧化物(ABO3)具有热稳定性高和氧迁移率好的特性,被认为具有良好的开发前景[8]。但钙钛矿型氧化物一般比表面积小,低温活性不佳,如沈柳倩[9]用共沉淀法制备的La0.8Cu0.2MnO3、La0.8Sr0.2MnO3和La0.8Mg0.2MnO3三种催化剂,甲苯的起燃温度在250 ℃左右,350 ℃时甲苯才可完全转化,无法满足冷启动时的尾气处理需求。Liu 等[10]研究制备了自熔聚合、共沉淀、溶胶−凝胶和浸渍方法的高活性锰钙钛矿型氧化物,当中使用自熔聚合法制备的钙钛矿型氧化物具有最好的催化能力,其甲苯转化率在270 ℃时保持在99.9%以上。为了高效激活催化剂的活性,CeO2常被作为催化剂活性载体,其储氧量十分丰富,Wang 等研究了La0.8Ce0.2MnO3负载在不同形态的CeO2上催化氧化甲苯的活性,发现负载在多面体形态的CeO2上效果最佳,在空速为12000 h−1条件下,240 ℃的甲苯转化率可达90%[11]。基于本课题组曾经有关负载型钙钛矿氧化NO 以及抗硫性能的研究,结果表明负载型钙钛矿拥有良好的NO氧化性能和抗硫性能[12],因此,本研究通过制备不同载体负载的LaCoO3/MO2(M= Zr、Ti、Ce)催化剂共同氧化甲苯与NO,期望实现非贵金属DOC催化剂深度氧化VOCs 的同时,部分氧化NO。
1 实验部分
1.1 催化剂的制备
载体的制备:CeO2的制备称取定量Ce(NO3)3·6H2O溶于100 mL 去离子水中,室温下中速磁力搅拌至完全溶解,同时加入配好的4 mol/L的NH3·H2O 溶液,直至pH = 10,持续搅拌3 h 后,室温抽滤冲洗至pH 恒定。得到的固体置于80 ℃烘干12 h,500 ℃焙烧4 h;ZrO2制备方法同上;TiO2使用中冶新材制备成品。
负载型钙钛矿通过柠檬酸溶胶-凝胶法制备,按照化学计量比计算并称量好相应的硝酸盐(ABO3中n(A)∶n(B) = 1∶1),混合溶于一定量去离子水中搅拌均匀;按物质的量比n(金属离子)∶n(柠檬酸)∶n(乙二醇) = 1∶2.5∶2.5,先称取一定量柠檬酸加入混合溶液中,升温到至50 ℃并搅拌30 min后;再加入乙二醇搅拌30 min;后加入相应载体粉末(质量分数w= 30%),再升温到80 ℃搅拌60 min;将混合溶液于80 ℃干燥箱干燥12 h,置于箱式电阻炉中600 ℃煅烧3 h,将样品研磨筛分取40−80 目备用。
1.2 催化剂的表征
XRD:在 日 本D/MAX-2500/PC 型 粉 末X 射线衍射仪上进行,在2θ= 10°−90°以 10(°)/min 的速率扫描。BET:采用美国麦克公司生产的MAC2920化学吸附仪测定各样品的比表面积测定。H2-TPR:使用美国麦克公司生产的MAC2920 全自动程序升温化学吸附仪,测试前将样品在氩气气氛下,300 ℃吹扫30 min,降温至80 ℃,然后气路切换为H2/Ar 混合气,从80 ℃升温至900 ℃进行还原,升温速率为10 ℃/min。XPS:采用美国赛默飞世尔的K-Alpha 型X 射线光电子能谱仪进行测定,C 1s校准结合能为284.8 eV。原位漫反射红外:实验使用美国赛默飞世尔的Nicolet iZ10 FTIR 光谱仪。实验前先将催化剂样品在300 mL/min N2中升温到400 ℃预处理30 min,冷却至250 ℃,通入不同气体成分并维持30 min后记录相应样品的光谱。
1.3 催化剂活性评价测试
使用固定床石英反应器,用0.6 g 催化剂(40−80 目)测定催化活性。原料气组成为:0.05%甲苯、0.05% NO(使用时)和6% O2,平衡气体为高纯度N2。总流量为300 mL/min,对应质量空速(WHSV)为30000 mL/(g·h)。进出口NO 的含量采用红外分析仪检测NO,复合气体分析仪GT-2000检测CO2,ppbRAE3000+仪器检测甲苯。测试温度为100−400 ℃。
使用下列方法分别计算NO 转化率(xNO)、甲苯转化率(xtoluene)和二氧化碳产率(wCO2),反应中NO 转化率(xNO)按下式计算:
式中,Nin为进口处NO 的体积分数,Nout为出口处NO 的体积分数。
反应中甲苯转化率(xtoluene)按下式计算:
式中,Tin为进口处甲苯的体积分数,Tout为出口甲苯的体积分数。
反应中二氧化碳产率(wCO2)按下式计算:
式中,Cout为出口处二氧化碳的体积分数,(Tin−Tout) × 7为甲苯完全燃烧时二氧化碳的体积分数。
2 结果与讨论
2.1 LaCoO3/MO2(M=Ti、Ce、Zr)催化剂单独氧化甲苯及NO 的性能研究
图1(a)展 示了LaCoO3与LaCoO3/MO2对甲苯的催化氧化性能。结果发现,钙钛矿型氧化物催化剂的氧化活性曲线符合“S”型,即随着温度从低温段(100−200 ℃)转化率低向高温变化时,甲苯转化率出现缓慢增加向突然剧烈增加的趋势。催化剂活性顺序为:LaCoO3/CeO2(t90= 245℃)>LaCoO3/ZrO2(t90= 285℃)>LaCoO3/TiO2(t90= 380℃)>LaCoO3(t90= 405℃),以上结果证实了负载型钙钛矿催化剂的催化活性总体比LaCoO3钙钛矿型氧化物有所增强。此外,不同载体的负载型钙钛矿催化剂的催化性能有明显差异,以CeO2为载体的LaCoO3/CeO2活性最佳。图1(b)为LaCoO3与LaCoO3/MO2在氧化甲苯时对应的CO2产率。图中随着温度的升高,CO2产率也逐渐增加。图中LaCoO3/CeO2表现出较高的CO2产率,在200 ℃时CO2产率可达70%,温度升高至250 ℃时基本实现最大的CO2产率(即100%),该结果明显优于ZrO2、TiO2负载的钙钛矿型氧化物。结果表明,LaCoO3/CeO2在表现出对甲苯最佳的催化氧化性能。
图1(c)展示了LaCoO3与LaCoO3/MO2催化氧化NO 的效果。与LaCoO3相比,各温度区间下LaCoO3/CeO2和LaCoO3/ZrO2的NO 转化率均明显提升,只有LaCoO3/ TiO2氧化NO 的活性没有明显改善。通常,快速SCR 反应比标准SCR 反应快10倍,当NO 与NO2的比例为1∶1 时,很好地满足了快速SCR 反应的发生[13]。以上结果证实,在实验条件下的LaCoO3/CeO2和LaCoO3/ZrO2催化剂可以提升DOC 性能,为后续的尾气处理提供良好条件。
2.2 LaCoO3/MO2(M=Ce、Zr)催化剂同时氧化甲苯和NO 的性能研究
根据活性实验证实了LaCoO3/CeO2和LaCoO3/ZrO2在250−400 ℃温度段获得了较高的甲苯与NO 的转化率,故进一步选用这两种负载型钙钛矿催化剂来考察NO 的存在是否会影响氧化甲苯的过程。图2(a)展示了LaCoO3/MO2(M= Ce、Zr)在有无NO 的情况下甲苯转化率变化。可以发现,存在NO 时LaCoO3/CeO2甲苯氧化的t90保持为245 ℃,并未受到NO 影响。但LaCoO3/ZrO2在300−400 ℃时甲苯的氧化活性受到了一定的抑制,当NO 存在时氧化甲苯的t90由285 ℃升高到350 ℃。CO2产率见图2(b),通入NO 后,LaCoO3/CeO2和LaCoO3/ZrO2在250 ℃以下CO2产率明显下降。进一步观察到随着温度升高,LaCoO3/CeO2则不受NO 影响,而LaCoO3/ZrO2依旧受NO 的影响。根据文献报道,这可能是由于NOx和甲苯时对活性氧的竞争吸附导致的[14,15]。
图2(c)展示了LaCoO3/CeO2、LaCoO3/ZrO2甲苯对NO 转化率的影响。在100−200 ℃低温段,在甲苯存在时,NO 的氧化受甲苯影响发生了失活现象。这是由于低温阶段甲苯氧化率低,甲苯及其副产物沉积在钙钛矿型氧化物表面,碳沉积很容易覆盖催化剂表面导致催化剂失活从而抑制了NO 的氧化活性[15,16]。随着温度的进一步升高,甲苯氧化率逐渐增加,催化剂表面不再被甲苯及副产物覆盖,因此,NO 的转化率也逐渐上升。从图2可以看出,当甲苯与NO 共同存在时,在275−350 ℃温度段LaCoO3/CeO2实现了甲苯的完全氧化,并且NO 依旧能够达到50%以上的转化率,从而为后续的DPF 再生和快速SCR 反应提供了有利条件。
2.3 LaCoO3/MO2 (M=Ti、Ce、Zr) 催化剂的表征
2.3.1 BET 和XRD 分析
对催化剂结构进行表征分析,有助于理解负载载体后催化剂的性能,从而为催化剂高效降解甲苯和NO 提供理论依据。表1 为LaCoO3、MO2载体和LaCoO3/MO2的比表面积的分析结果。可以发现,LaCoO3被CeO2、ZrO2后,比表面积明显增加。例如,LaCoO3/ZrO2和LaCoO3/CeO2比表面积从6.3 m2/g 分别增加至37.9、41.6 m2/g。钙钛矿型氧化物的比表面积越大,可为活性组分提供更多的接触位点,从而有利于氧化活性的提高[17]。

表1 催化剂的比表面积Table 1 Specific surface area of catalysts
由LaCoO3和LaCoO3/MO2的XRD 谱图(图3)中发现,CeO2、TiO2、ZrO2分别为方体萤石结构、四方晶系锐钛矿型和单斜相晶型[18−20]。此外,在LaCoO3/MO2催化剂上可检测到LaCoO3的特征峰,说明溶胶-凝胶法可成功制备出LaCoO3晶型并负载在载体上。但LaCoO3的特征峰均为微弱衍射峰,表明活性组分均匀地分散在载体上或形成微晶而并未形成明显的LaCoO3晶相结构。
2.3.2 H2-TPR 表征
为了探究催化剂的氧化还原能力,对LaCoO3与LaCoO3/MO2进行H2-TPR 表征,图4 呈现了催化剂的耗氢量和起始还原温度。在LaCoO3中,300−500 ℃所对应的是Co3+→Co2+双肩峰,这意味着存在两种状态Co3+物种,较低温峰为活性氧附近的Co3+发生了还原反应,而较高温峰为晶格中Co3+的还原;500−700 ℃对应的还原峰为 Co2+→Co0。与LaCoO3相比,由于活性组分的减少,LaCoO3/MO2的Co 还原峰,强度与耗氢量均显著减少;另外发现,由于LaCoO3活性组分均匀分散,有利于Co 离子的还原,Co3+→Co2+还原峰明显向低温方向移动,该还原峰是影响甲苯与NO 氧化的关键[21]。此外,LaCoO3/CeO2和LaCoO3/ZrO2的Co3+→Co2+还原峰为单峰形式,这意味着活性氧附近的Co3+数量增加,从而证实了对甲苯和NO 的高效氧化。特别是LaCoO3/CeO2在400 ℃内耗氢量最大,从而证实了LaCoO3/CeO2在催化甲苯和NO 时表现出更好地活性。
2.3.3 XPS 表征
进一步对反应前后LaCoO3与LaCoO3/MO2的O 1s、Co 2p以及相应载体Zr 3d、Ce 3d、Ti 2p进行了XPS 的表征分析,以探究催化剂表面相的元素组成(元素类型及价态)及比例。表2 列出了钙钛矿型氧化物主要元素在催化剂表面成分及各自的Olat和Oads的结合能,LaCoO3的O 1s分别在528.8、531.2 eV 附近出峰;如结合图5 分析,LaCoO3/MO2吸附氧的结合能无明显改变,而晶格氧的结合能在529.4 eV 附近出峰,LaCoO3/MO2有着更高的晶格氧结合能,晶格氧更加活跃[22]。根据表2 可知,钙钛矿型氧化物Oads/Olat的比值大小为LaCoO3>LaCoO3/CeO2> LaCoO3/ZrO2> LaCoO3/TiO2。反应后,负载型钙钛矿Oads/Olat比值均减小,表明Oads参与了反应,其中,LaCoO3/CeO2比值明显下降,说明形成了更多氧空位,从而促进反应过程中气态氧的吸附和晶格氧的流动;而LaCoO3的比值有所上升,可能是在催化反应过程中气相氧补充氧空位而引起的[23]。
在Co 2p的XPS 中进一步发现了多价态金属Co,Co 2p3/2和Co 2p1/2的结合能分别位于780、795 eV,钙钛矿型氧化物中779.4−779. 7 eV 与794.4−794.6 eV的峰为Co3+的峰,781.4−781.7 eV 和795.9−796.2eV处的峰为Co2+的峰。这表明LaCoO3与LaCoO3/MO2中存在Co2+、Co3+。根据电中和原理,Co2+的存在会诱导形成氧空位,促进气态O2向Oads的转变,从而为催化反应提供更多的活性位点[21]。此外,在Zr 3d、Ce 3d、Ti 2p的XPS 分析中,元素Zr 与Ti 未发生价态变化[24,25],而Ce 元素存在多价态,通常,Ce 3d由八个3d电子相关的峰组成,v、v′、v′′、v′′′为Ce 3d5/2电离,u、u′、u′′、u′′′为Ce 3d3/2,其中,v、u'为Ce3+的峰,其余为Ce4+的峰。当Ce3+与Ce4+的共同存在时,会促进了化学氧化还原过程循环[24]。考虑到LaCoO3/CeO2中存在Ce3+、Ce4+、Co2+、Co3+离子,而表2 中 LaCoO3/CeO2反应前后Co3+/Co2+与Ce3+/Ce4+的比值增加,这表明 LaCoO3/CeO2在反应过程中发生了Co2++Ce4+↔Co3++Ce3+氧化还原循环反应,即在钙钛矿型氧化物与载体之间存在着相互作用,造成晶格氧的缺陷,这有利于氧空位的形成[14,26,27]。
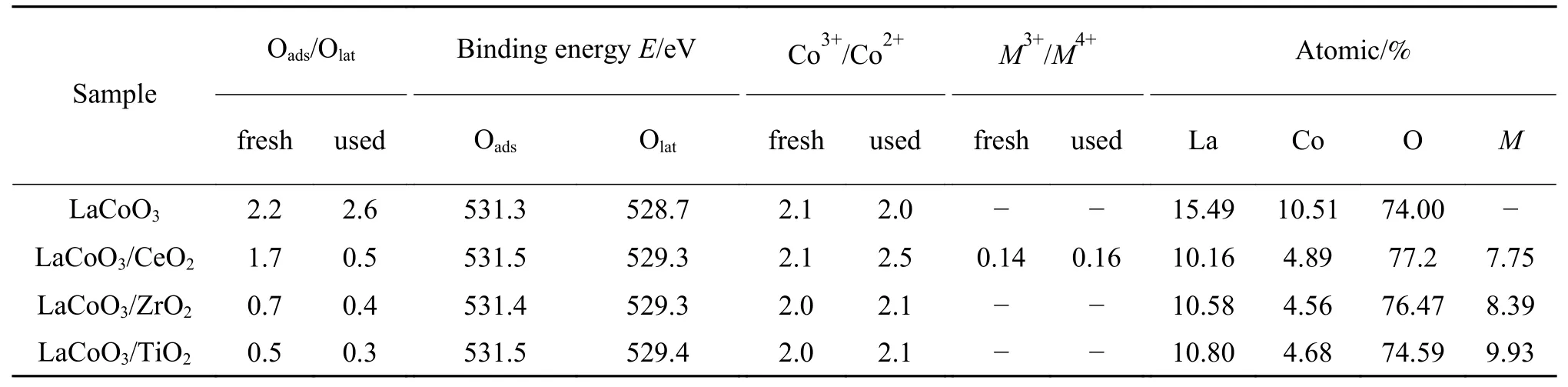
表2 催化剂的XPS 表面元素比值以及结合能Table 2 XPS results of surface element ratio and binding energy
2.3.4In-situDRIFTs 分析
利用原位漫反射红外研究反应中间体的形成,进一步阐明LaCoO3/CeO2催化剂对甲苯及NO氧化的反应机理。图6(a)展示了250 ℃下随时间变化的甲苯吸附和甲苯+NO 共吸附的DRIFT 谱图。3068−2872 cm−1处的特征峰归属于芳香环C–H的伸缩振动,1600−1300 cm−1处为芳香环C = C 的拉伸振动,1390 cm−1为不对称/对称甲基的弯曲振动[27,28]。在甲苯单独通入时,这些特征峰强度在短时间内快速增强,这表明LaCoO3/CeO2对甲苯有较强的吸附能力。1547 cm−1处形成较强的吸收峰属于羧酸基团,表明甲苯吸附过程中催化剂表面形成了苯甲酸盐物种。而在1338、1330、1147、1078 、1240 cm−1吸收峰归属于具有Ph–CH2–O–结构的苯甲醇物种[15,27,29],这表明,LaCoO3/CeO2活跃的晶格氧具有将甲苯氧化为羧酸盐、苯甲酸盐物种、苯甲醇等关键中间氧化产物的能力。NO+甲苯共吸附时,1494 cm−1处出现了单齿硝酸盐的特征峰,同时1596、1455、1390 、1547、1338 cm−1处的峰有一定程度地减弱,这表明,NO 与甲苯吸附产生的中间产物发生反应。为探究O2对甲苯氧化以及NO 存在时甲苯的氧化影响,图6(b)展示了向预吸附甲苯和甲苯+NO 的LaCoO3/CeO2催化剂通入O2后的DRIFT 谱图。O2通入后,1547、1455、1390、1338 cm−1的吸收峰迅速减弱,同时在2331 cm−1处出现了CO2的特征峰[30],表明甲苯以及中间氧化产物被O2氧化为了CO2和H2O。而当O2通入至预吸附甲苯+NO 的LaCoO3/CeO2催化剂时,1494、1393 cm−1处吸收峰强度的增加表明NO 与O2生成了硝酸盐物。同时1946、1908、1810 cm−1处出现比较明显的酸酐的不对称/对称的υ(C = O)伸缩振动,而1574、1455、1338 cm−1处的吸收峰强度随着反应的进行明显减弱,这表明甲苯在NO 共吸附时有较好的氧化性能。
图6(c)展示了O2通入的条件下LaCoO3/CeO2催化剂上甲苯单独氧化后加入NO 共同氧化的DRIFT 谱图,可以观察到单独氧化甲苯时芳香环、甲基、苯甲醇、苯甲酸的谱带随着时间的延长明显增强,但值得注意的是在1946、1908、1810 cm−1处出现酸酐的不对称和对称υ(C = O)拉伸振动[27],表明甲苯及氧化中间产物在催化剂上不断堆积。在甲苯氧化30 min 后加入NO 共同氧化时,可以观察到甲苯以及中间氧化物的特征峰持续增强,但未观察到有关NO 的明显谱带,这表明NO 的通入不影响甲苯及其关键中间产物的生成。图6(d)展示了NO 单独氧化以及加入甲苯共同反应的DRIFT 谱图。在单独氧化NO 时,通入5 min 后1593 cm−1处出现NO2吸附峰[31],但随着反应的进行而消失,同时1494、1353、1249 cm−1处吸收峰逐渐增强,其中,1494、1249 cm−1为单齿硝酸盐的特征峰,1353 cm−1处为亚硝酸盐的特征峰,这表明NO 主 要 吸 附 在Brønsted 酸 位 上 形 成但通入甲苯时,1249、1353 cm−1的吸收峰强度随着时间推移明显下降,而有关甲苯在1596、1547、1390、1330、1078、3068−2872 cm−1的特征峰逐渐增强,特别是苯甲醇、苯甲酸、甲基的吸收峰。
2.4 LaCoO3/CeO2 的反应机理
根据表征分析结果,LaCoO3/CeO2催化剂上NO 和甲苯的反应机理如下。
NO 的氧 化:NO 首 先 会被吸 附 在Brønsted 酸位点上,与催化剂上的活性氧反应生成硝酸盐和亚硝酸盐,反应符合Langmuir-Hinshelwood 机制:
甲苯的氧化:甲苯上的甲基与催化剂表面的吸附氧发生相互作用,使甲苯吸附在催化剂上,经过一步脱氢形成苯甲醇物种后,随后发生一个快速脱氢反应,形成苯甲酸盐物种,后进一步被氧化为CO2和H2O,甲苯的氧化为Mars-van Krevelen机制:
3 结 论
本研究制备了一系列负载型钙钛矿催化剂并探究其催化氧化甲苯与NO 的性能。结果表明,在以CeO2作为载体的LaCoO3/CeO2催化剂的甲苯与NO 催化氧化性能得到明显提升,甲苯的t90为245 ℃, NO 转化率在300 ℃时可达68%。LaCoO3/CeO2拥有更大的比表面积,具有更活跃的晶格氧和更好的氧化还原性能。同时LaCoO3/CeO2催化剂上Co 离子与Ce 离子存在电子转移,这有利于氧空位的形成从而增加了活性氧的迁移,促进了甲苯和NO 的氧化。在LaCoO3/CeO2催化剂上NO对甲苯的氧化未表现出抑制作用,这是由于NO与甲苯吸附产生的中间产物发生反应。此外,原位红外漫反射进一步揭示出在LaCoO3/CeO2催化剂上甲苯和NO 的氧化分别符合Mars-van Krevelen机制和Langmuir-Hinshelwood 机制。
猜你喜欢
无机化学学报(2022年8期)2022-08-09
盐科学与化工(2022年6期)2022-06-20
可再生能源(2022年5期)2022-06-09
城市道桥与防洪(2022年3期)2022-05-08
无机材料学报(2022年1期)2022-04-12
燃烧科学与技术(2021年5期)2021-10-28
能源工程(2021年2期)2021-07-21
湖北农机化(2021年11期)2021-07-01
安全与环境工程(2021年2期)2021-04-02
分析化学(2019年3期)2019-03-30
