
3 例Lesch-Nyhan 综合征患儿及其父母/姐姐基因检测分析
2022-07-29 03:02岳璇刘晓鸣刘莉仇莉陈曼陈娇
山东医药 2022年18期
岳璇,刘晓鸣,刘莉,仇莉,陈曼,陈娇
徐州市儿童医院神经内科,江苏徐州 221006
Lesch-Nyhan 综 合 征(Lesch-Nyhan syn‐drome,LNS)是由次黄嘌呤-鸟嘌呤磷酸核糖基转移酶(Hypoxanthine-guanine phosphoribosyltransferase,HGPRT)基因突变导致。LNS 是一种罕见的X连锁隐性遗传病,在欧洲发病率为1/40 万~1/20 万,在中国人群中发病率极低。主要临床表现为运动、智力发育落后、运动障碍、尿酸增高及自残等症状。因早期临床症状常以运动、智力发育落后为主要表现,不具有特异性,常被临床误诊为脑性瘫痪,对症康复治疗后症状未得到改善,后期患儿可能出现咬手、脚等自伤行为。目前我国对LNS 的相关研究报道较少,临床医师普遍对LNS的认识不够,导致LNS的早期诊断困难。我们近期收治了3 例Lesch-Nyhan 综合征患儿,现分析患儿及其父母/姐姐的基因检测信息,总结其遗传特征。
1 资料与方法
1.1 临床资料 选择徐州市儿童医院神经内科收治的3 例Lesch-Nyhan 综合症患儿。患儿的治疗均经监护人知情同意签字,本研究经医院伦理及医疗技术管理委员会批准同意。
例1,男,2岁11个月,因“咬手、咬嘴10 个月”于2021年5月就诊于我院。患儿自幼发育落后,6月龄竖头,1 岁2 个月时可独坐数秒,至入院时仍不能扶站及扶走,发“爸爸、妈妈”2 个字叠音词,一直在当地医院按照“脑性瘫痪”予康复治疗,2岁1个月时患儿出现不自主咬手指、咬嘴症状。患儿系第2胎,第2产,足月顺产,否认围生期缺氧窒息史。例2,男,5岁,为例1同胞哥哥,自幼运动发育落后,于当地医院诊断脑性瘫痪,一直进行脑性瘫痪康复治疗,后出现咬手动作。患儿无出生时缺氧窒息病史,2 岁时曾因肾结石行手术治疗,既往视频脑电图检查为正常脑电图,MRI检查未见异常。父母体健,非近亲婚配,患儿外公、外婆健康。入院检查结果为体质量12 kg,发育落后,下唇破溃,局部化脓,双手指破溃,四肢肌张力高,能独坐数秒,不能扶站及扶走。入院时生化检查可见尿酸575 μmol/L(正常值90~420 μmol/L)。视频脑电图(2 岁11 个月)为界限性脑电图,背景活动偏慢,指认事件不伴发作期图形。头颅磁共振成像(MRI)检查未见异常。Gesell发育评估应人能48.0 分,应物能48.0 分,粗大动作48.0 分,精细动作30.0 分,言语能48.0 分,发育商44.4分。
例3,男,3 岁1 个月,因“发育落后,自咬嘴唇半年”入院,患儿为第3胎第3产,足月剖宫产,出生时无产伤及窒息史,生后发现患儿吸吮无力,3月龄发现竖头不稳,现不能独坐,不能站立,双眼能追视,仅能叫“爸爸、妈妈”,近半年出现咬嘴唇,手指等自残行为,同时发现小便颜色发白。父母体健,非近亲婚配,患儿爷爷、奶奶及外公、外婆健康。入院后查体:肌张力高。入院后查尿酸486 μmol/L,血常规、肝肾功正常,头颅MRI检查未见异常。脑电图检查为正常脑电图。
1.2 受检对象基因全外显子组基因检测及分析征得患儿监护人知情同意后,采集患儿及其家系(例1、例2、例1 父母、例3、例3 大姐、例3 二姐及例3 父母)的外周静脉血,送至武汉康圣达医学检验所及智因东方公司,对患儿及其家系外周血DNA 进行全外显子组高通量测序。测序结果经HGMD 库和Clin‐var 库及gnomAD 库检索对比,异常结果通过美国医学遗传学与基因组学学会(The American College of Medical Genetics and Genomics,ACMG)基因突变分级,采用进化保守性预测软件SIFT,蛋白结构预测软件Polyphen2,蛋白结构预测软件MutationTaster分析致病性变异。突变基因位点进行致病变异分级,PM1 为错义变异位于深入研究的无良性变异的外显子功能域(clinvar 无benign 变异),已确定的致病变异的错义变异且位于致病热点区;PM2 为正常对照人群数据库中均未发现,属于低频变异;PM5为错义变异位于先前已确定为致病性的氨基酸残基;PM8为多种统计方法预测出变异对基因(基因产物)有影响;PP4 为基因关联疾病与先证者表型高度相符。通过Sanger法验证突变位点并对患儿进行家系分析。
2 结果
例1 在X 染色体chrX 转录本133609312 存在HPRT1 基因c.236T>C,为致病半合子突变,即该变异的染色体位置为chrX:133609312(基因组版本:hg19),转录本NM_000194,位于第3 个外显子,导致编码区第236 号核苷酸T 突变为C,导致第79号氨基酸由亮氨酸(leu)变为苯丙氨酸(Phe)。其父亲(表型正常)该位点基因为野生型,母亲(表型正常)与例1 存在同一位点突变,为杂合突变,家系验证突变来源于母亲。参考ACMG 基因突变解读指南,例1 突变位点在HGMD 专业版数据库中无收录,Clinvar 数据库中无收录。该位点的人群频率无收录。在gnomAD 数据库的东亚人群中无收录。SIFT 软件预测D(评分:0.002),Polyphen2 软件预测为D(评分:0.998),MutationTaster 预测D(评分:1.0),说明基因关联疾病与先证者表型高度相符。该位点突变符合PM1、PM2、PP3 及PP4,致病变异分级为可能致病。例2在X染色体chrX:133609312存在HPRT1 基因c. 236T>C,为致病半合子突变。例1、2 及其父母X 染色体HPRT1 基因c. 236T>C 基因序列见图1~4。
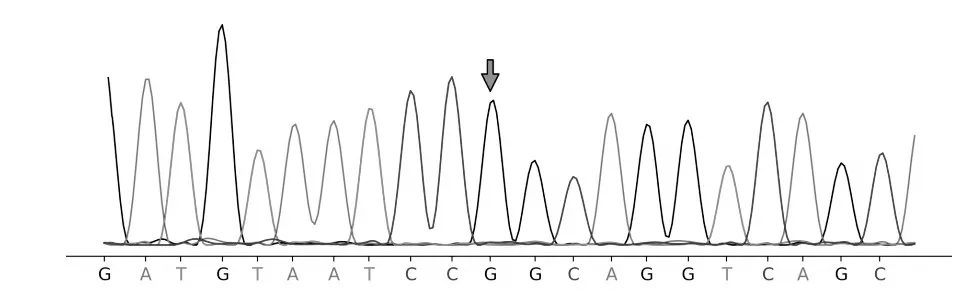
图1 例1的X染色体HPRT1基因c.236T>C基因序列
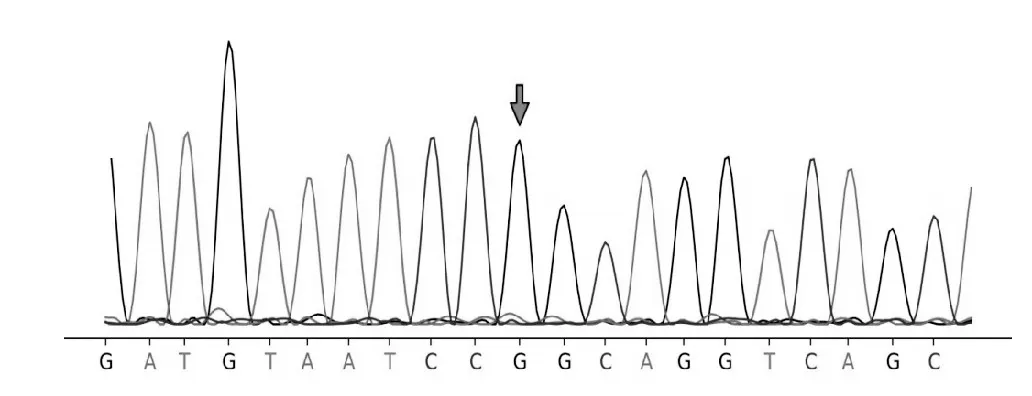
图2 例2的X染色体HPRT1基因c.236T>C基因序列
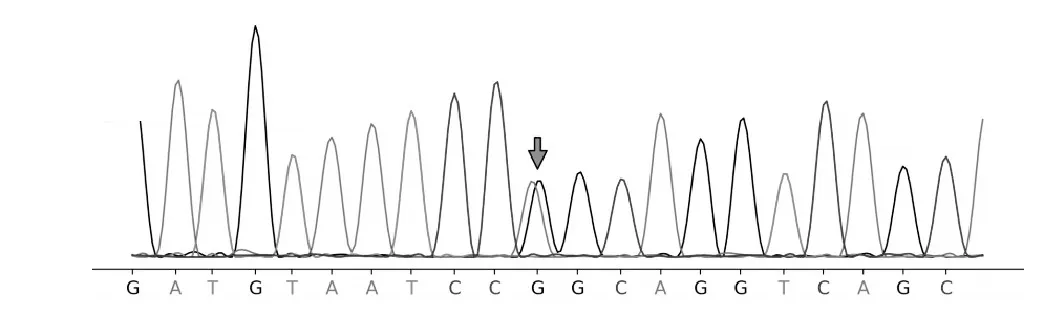
图3 例1、2母亲的X染色体HPRT1基因c.236T>C基因序列
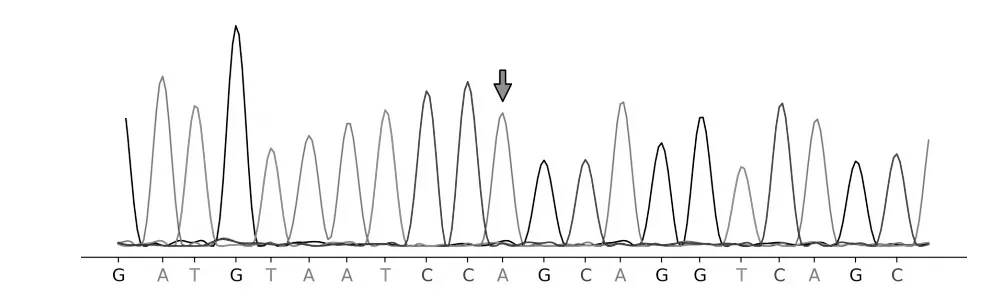
图4 例1、2父亲的HPRT1基因序列
例3 在X 染色体上3 号外显子存在HPRT1 基因c. 197G>A,为致病半合子突变,编码区第197 核苷酸G突变A,导致第66号氨基酸由半胱氨酸cys变为甘氨酸Gly。父亲及二姐(表型正常)该位点为野生型,母亲及大姐(表型正常)该位点均为杂合子;与例3 有同一位点突变,为杂合突变,家系验证例3 突变位点来源于母亲。致病变异分级评定该突变位点符合PM2、PM5、PM8 及PP4,致病变异分级为可能致病。
3 讨论
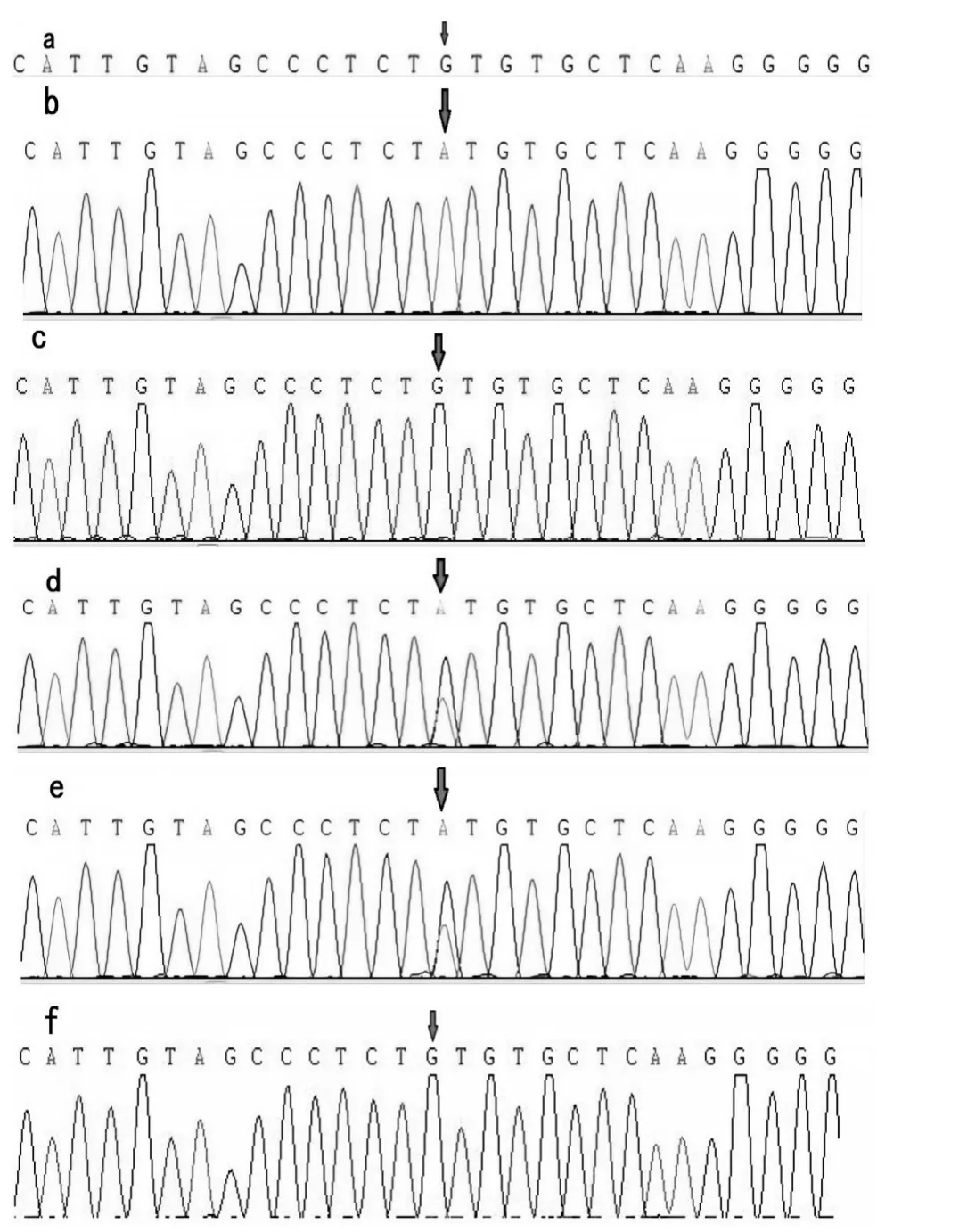
图5 例3及其父母姐姐的HPRT1基因序列
Lesch-Nyhan 综合征是一种罕见病,在欧洲的发病率为1/40 万~1/20 万[1-2]。Lesch-Nyhan 综合征是一种X连锁隐性遗传性疾病,患者均为男性,女性携带者不发病。HPRT1 基因是Lesch-Nyhan 综合征的惟一致病基因,其位于X 染色体q26.3 区域,全长40.5 kb,包含8 个内含子和9 个外显子,编码含有218 个氨基酸。次黄嘌呤-鸟嘌呤磷酸核糖转移酶(HGPRT)可把次黄嘌呤转化为次黄苷酸,把鸟嘌呤转化为鸟苷-磷酸[3]。次黄嘌呤及鸟嘌呤的嘌呤基团代谢为尿酸。而人体内嘌呤核苷酸的合成有两种途径:从头合成途径以及补救合成途径[4]。脑组织缺乏重头合成途径的酶体系,只能进行补救合成途径,而HGPRT 是补救途径中的关键酶,因此HPRT1基因异常既能导致高尿酸血症,也可能影响神经系统的功能[5]。根据HGPRT 缺失程度差异,临床主要分为3 型:①经典型Lesch-Nyhan 综合征。HGPRT酶活性<1.5%,临床表现为代谢及神经系统异常,高尿酸血症,发育迟缓,肌张力障碍、脑性瘫痪及自伤行为。②中间型。HGPRT 酶活性为1.5%~8.0%,临床表现为代谢及神经系统异常,但神经系统受累较轻,不伴自伤症状。③HGPRT 相关高尿酸血症。HGPRT 酶活性>8.0%,临床仅表现为高尿酸血症,不伴神经系统症状。HGPRT 完全缺失者,发病早,病情进展迅速,致残率高。
Lesch-Nyhan 综合征患儿通常在出生半年内出现运动发育落后,肌张力低下,后逐渐出现痉挛性瘫痪、腱反射亢进等锥体束症状以及肌张力障碍及舞蹈徐动症等锥体外系表现。临床常将Lesch-Nyhan综合征误诊为脑性瘫痪后进行脑性瘫痪康复治疗。本研究中例3 例患儿无围产期高危因素,出生时无缺氧窒息病史,父母既往体健,出生后3例患儿均出现发育落后,后出现肌张力障碍,被临床认为脑性瘫痪,一直康复治疗。例2既往发现多次尿酸增高,但始终未引起重视,后出现小便异常,行肾脏彩超发现结石,予结石手术治疗,病因一直未明确,说明本病早期症状特异性不高,临床早期识别存在困难。其次患儿自伤行为是本病较为特征的临床表现,一般在1 岁后出现,3 岁时自伤行为变得明显,出现自我咬伤口唇、舌、牙齿、手指、肩部、前臂,有时会撞击头部和肢体等,严重时可导致牙齿脱落,手指节断裂。例1 患儿约2 岁1 个月出现自伤行为,例2 自伤行为患儿母亲未及时发现,病例1确诊后,患儿母亲才发现例2 存在自残行为。但是自残行为如何出现,具体发病机制尚不明确。
目前文献及数据库报道的HPRT1 基因突变位点有600 多个。HPRT1 基因的突变方式多种多样,有错义突变、无义突变、剪接位点突变、缺失及重复等。有学者[6]通过大样本数据分析,提出HPRT1 编码区的缺失突变是常见突变类型,剪接突变其次,无义突变常引起LNS,重复突变较少引起较轻的临床表型,错义突变多引起其变异型,表现为Lesch-Nyhan综合征、HPRT相关的高尿酸血症(HRH)以及神经系统异常综合征(HRND)等。外显子3 可能为HPRT1 基因的突变热点[7],外显子3 编码的氨基酸属于HPRT 蛋白的CpG 岛区域,而CpG 岛区域是一些基因的重要调控区域[8],因此该区域的突变可导致酶活性完全丧失,临床表现为Lesch-Nyhan 综合征。本文3例患儿突变均发生在3号外显子,为错义突变,患儿临床表现与基因所致经典型Lesch-Nyhan综合征一致,例1、2 发现HPRT1 基因位于X 染色体上(转录本133609312),3 号外显子(转录本NM-000194),发现C. 236T>C 的核苷酸变异。例3 发现X 染色体上3 号外显子c.197G>A 的核苷酸变异,上述变异分别导致第79号氨基酸由leu变为Phe,66号氨基酸由cys 变为Gly。先证者其母上述位点为杂合子,该变异可能导致蛋白功能受损,与LNS 相关。上述变化均不属于多态性变化,在人群中发生的频率极低。HPRTl 基因是LNS 的致病基因,为X 连锁隐性遗传方式。对于该类遗传方式,所发现的变异可能导致发病。在例1、2、3外周血HPRT1基因所发现的变异遗传均来自受检者母亲,其母为杂合子,符合X 连锁隐性遗传方式。本研究中例1 有尿酸高症状,2岁1个月出现自残行为,而例2发现4岁后出现自伤行为,2 岁左右已经出现肾结石行手术治疗,例3 有尿酸高及自残行为,例1、2 为同一位点基因变异,3 例患儿均为错义突变,患儿临床表型不完全相同。因此,LNS 病情严重程度除了与HPRTl 基因突变位点存在密切联系,还与患儿体内HGPRT酶活动水平相关。
目前对于Lesch-Nyhan 综合征导致神经系统症状的具体作用机制不清楚,临床只能根据患儿症状进行对症治疗。表现为高尿酸血症的患儿应少食动物内脏、海鲜等,以低嘌呤饮食为主。可采用人为约束、保护牙齿、口唇及手指等预防患儿的自伤行为,针对肌张力障碍,选用巴氯芬、苯二氮类药物,深部电刺激等。
综上所述,Lesch-Nyhan 综合征患儿主要临床表现为发育落后、肌张力异常及自伤行为等。Lesch-Nyhan 综合征患儿存在HGPRT 基因chrX:exon3 c. 236T>C,p. L79P、chrX exon3c. 197G>A,p.C66Y 突变,致病变异分级为可能致病,均遗传自患儿母亲。存在发育落后、肌张力障碍合并尿酸升高、自伤症状的患儿,应警惕Lesch-Nyhan 综合征可能。
猜你喜欢
电子科技大学学报(2022年5期)2022-10-29
中国社区医师(2022年14期)2022-06-10
广西医科大学学报(2022年5期)2022-06-07
中国循证儿科杂志(2022年2期)2022-05-26
中国典型病例大全(2022年9期)2022-04-19
昆明医科大学学报(2022年3期)2022-04-19
今日健康(2021年12期)2021-11-30
中国生殖健康(2020年4期)2021-01-18
山东医药(2020年32期)2020-12-31
中南医学科学杂志(2019年6期)2019-12-05
