
猪SALL1蛋白分子信息分析及基因敲除细胞系制备
2022-07-16 04:59李美双赵丽华李荣凤
南京医科大学学报(自然科学版) 2022年7期
李美双,赵丽华,李荣凤,*
1南京医科大学生殖医学国家重点实验室,2江苏省异种移植重点实验室,江苏 南京 211166
肾脏疾病是最常见的疾病之一,全球约有8.5 亿患者饱受急性或慢性肾衰竭的折磨[1]。目前由于透析设备以及肾脏供体的短缺,每年约有5 万人死于肾衰竭[2]。肾脏透析疗法虽然可以代替肾脏完成排泄功能,但是并不能代替肾脏行使内分泌功能。肾脏移植虽然在一定程度上恢复肾脏功能,但也由于器官供应的短缺和免疫抑制药物的相关问题而受到阻碍,因此开发新的低免疫原性的器官具有较大的应用前景。利用患者来源的干细胞通过囊胚互补技术,有望在大动物体内再生人源化的肾脏器官。囊胚互补首先利用基因编辑技术在胚胎发育早期阶段敲除决定某个器官发育的关键基因,然后待胚胎发育到囊胚阶段后将多能干细胞注入到囊胚腔内,干细胞将参入胚胎形成嵌合体,并优先去形成由于关键基因敲除导致的缺失的器官。Chen 等[4]将正常小鼠胚胎干细胞注射到来自rag-2缺陷小鼠的囊胚中,获得了其成熟B 细胞和T 细胞均来源于供体干细胞的嵌合小鼠。此后,研究者们对囊胚互补的研究逐渐深入,胸腺[5]、甲状腺[6]、肺[6-7]等脏器的种内囊胚互补均已出现。2010 年Kobayashi 等[8]利用囊胚互补技术在小鼠体内生成大鼠胰腺,论证了利用囊胚互补进行异种器官生成的可行性。这提示研究者去探索在人类以外的物种中产生具有功能的人类器官。考虑到在解剖学、生理学等方面与人类的相似之处,猪被人认为是再生人源性器官的合适载体[9]。
CRISPR 即规律间隔成簇短回文重复序列,最早于1987年由Ishino在大肠杆菌中发现。研究者们将CRISPR 与Ⅱ型CAS 蛋白相结合,使其迅速成为应用最广泛的基因编辑工具[10]。体细胞核移植与CRISPR/Cas9 的联合应用已广泛用于大动物基因编辑模型的构建[11]。例如,2017 年Zhang 等[12]将C3基因敲除,产生了血浆中C3蛋白缺乏的猪模型;2020年Fischer等[13]获得了GGTA1/CMAH/B4GALNT2三基因敲除的猪模型用于异种免疫排斥的研究。这说明结合体细胞核移植技术与CRISPR/Cas9 技术获得肾脏发育缺陷猪模型的可能性。2013 年Matsunari等[14]首先获得了Pdx1-/-胰腺发育缺陷的猪模型,随后他们将带有荧光标记的另一品系的猪胚胎卵裂球注入Pdx1-/-囊胚中,获得了来源于荧光标记细胞的具有正常组织特征及功能的外源胰腺。2020年Matsunari 等[15]又在HHEX-/-肝脏缺陷的猪模型中通过种内囊胚互补获得了肝脏发育正常的猪胎儿,这证明了猪进行种内囊胚互补的可能性。此外,2017 年Wu 等[16]提出人类干细胞经囊胚互补可移植并分化为猪胚胎中的多个谱系。2020年Das等[17]将人诱导多能干细胞注射入ETV-/-猪囊胚中,其后代的血管内皮细胞均来源于人类细胞。因此,本研究认为通过CRISPR/Cas9基因编辑技术获得肾脏器官发育缺陷的猪模型,并将其与囊胚互补技术结合,或许可以实现在猪体内再生人的肾脏器官。
Spalt 样转录因子1(spalt like transcription factor1,SALL1)在肾脏发生过程中极为重要。Sall1基因主要表达于输尿管芽周围的生后肾原基内,参与诱导输尿管芽反复分支形成输尿管、肾盂、肾盏和集合小管的肾脏发育过程。2001 年Nishinakamura等[18]研究发现Sall1基因的纯合缺失小鼠在出生后24 h 内死亡,表现出肾发育不全或严重发育不良。2019 年Goto 等[19]利用囊胚互补技术将大鼠胚胎干细胞注射到Sall1敲除小鼠囊胚内,嵌合体比例为69.1%,这提示通过敲除猪Sall1基因获得肾脏发育空位,从而进行人猪异种囊胚互补的可能性。
关于猪肾脏发育不良模型,虽然Watanabe等[20]进行了研究,但其获得的动物模型依然存在一定的局限性,目前为止尚未见到肾脏完全缺失的猪模型的报道。本研究通过对人、猪、鼠SALL1 蛋白的同源性进行分析,进一步验证了猪与人SALL1蛋白的相似性,同时通过CRISPR/Cas9 基因编辑技术对巴马小型猪胎儿成纤维细胞进行Sall1基因敲除,为获得肾缺失猪模型以及研究Sall1基因在猪肾脏发育过程中的作用提供研究材料,并为下一步获得肾缺失模型以及进行人源性干细胞囊胚互补奠定基础。
1 材料和方法
1.1 材料
35 d 猪胎儿成纤维细胞由本实验室制备并培养。DMEM 培养液、DPBS、0.25%Trypsin-EDTA、G418 和Penn/Strep solution(Gibco 公司,美国),胎牛血清(Sigma 公司,美国),PX330 骨架质粒(Addgene公司,美国),BbsⅠ核酸内切酶、Quick Ligase连接酶(New England Biolabs 公司,美国);FastPure Gel DNA Extraction mini Kit(南京Vazyme 公司),无内毒素质粒大提试剂盒(北京TIANGEN 公司),P3 细胞核转染试剂盒(Lonza 公司,德国),DH5α感受态、pMD 18T载体(TaKaRa公司,日本)。
1.2 方法
1.2.1 人、猪、鼠SALL1蛋白的生物信息学分析
下 载NCBI(https://www.ncbi.nlm.nih.gov/pmc/)中所提供的人、猪、鼠SALL1蛋白的氨基酸序列,使用DNAMAN 软件比对分析人与猪SALL1 氨基酸的相似度及人与鼠SALL1氨基酸的相似度;使用在线网站SOPMA(https://npsa-prabi.ibcp.fr/)比对分析人、猪、鼠SALL1 蛋白的二级结构,预测其α螺旋、β折叠、β转角和无规卷曲的比例;使用在线网站SWISS MODEL(https://www.swissmodel.expasy.org/)预测人、猪、鼠SALL1 蛋白的三级结构,再使用PyMOL 软件比较人与猪及人与鼠SALL1 蛋白三维结构的相似度;使用NCBI中CD-search工具比对人、猪、鼠SALL1蛋白的保守结构域。
1.2.2 sgRNA的设计与CRISPR/Cas9载体构建
根据NCBI 中所提供的猪Sall1基因序列(Gene ID:100519899),在第1外显子两端设计可用于扩增Sall1基因的PCR 引物并送至南京金斯瑞生物公司合成。以巴马小型猪胎儿成纤维细胞(porcine fetal fibroblast,PFF)基因组DNA 为PCR 模板,反应条件为:95 ℃5 min;95 ℃30 s,64 ℃30 s,72 ℃1 min,35 个循环;72 ℃7 min。通过琼脂糖凝胶电泳获得单一条带后将产物送至生物公司测序,确定巴马小型猪中Sall1基因的具体序列。根据测序获得的Sall1基因序列,利用CRISPR 在线设计网站(http://crispor.tefor.net/),遵循靶点设计原则(5′端为G,3′端为PAM序列,即NGG)选择合适的靶点位置,设计长度为20 bp的sgRNA 序列并在5′端进行磷酸化修饰。
CRISPR/Cas9 载体构建由PX330 载体线性化、引物退火、PX330 线性化载体与退火引物连接等步骤组成。首先使用BbsⅠ核酸内切酶酶切PX330 质粒使其线性化,同时将合成的5′磷酸化的sgRNA 序列于37 ℃孵育30 min,95 ℃孵育5 min,然后将其以5 ℃/min 的速率将温度降至25 ℃。再使用Quick Ligase连接酶将已线性化的PX330质粒与退火引物连接,反应条件为25 ℃连接10 min。最后将连接产物转化至大肠杆菌感受态DH5α中,挑取单菌落保存并送测序公司检测sgRNA 是否成功连入PX330质粒,测序引物为:5′-ACTATCATATGCTTACCGTAAC-3′。经测序分析后选择连接成功的菌液使用大提质粒试剂盒提取质粒。
1.2.3Sall1基因PX330-sgRNA载体打靶效率验证
使用DMEM 培养基(含15%胎牛血清)于6 cm培养皿中培养原代PFF 细胞,待细胞融合度达到90%左右时,0.05%胰蛋白酶消化并收集细胞。细胞计数后取1×106个细胞,按照P3 细胞核转染试剂盒说明书,利用程序EN150,将5 μgSall1-PX330-sgRNA质粒转入细胞中。培养48 h 后提取细胞基因组。PCR 扩增打靶区域后进行琼脂糖凝胶电泳,切胶回收纯化DNA,并用测序分析方法检测sgRNA的打靶效率。首先,PCR 扩增打靶区域基因组后送至南京思普金生物科技有限公司测序,PCR 引物和反应条件同1.2.2。然后,以野生型细胞的测序结果为对照,利用在线分析软件(https://tider.deskgen.com/)分析打靶载体的效率。
1.2.4 细胞转染和单克隆细胞的筛选鉴定
使用DMEM 培养基(含15%胎牛血清)于6 cm培养皿中培养原代PFF 细胞,待细胞融合度达到90%左右时,0.05%胰蛋白酶消化并收集细胞。将8 μg 的Sall1-PX330-sgRNA 打靶载体与1 μg 的Td-tomato(G418抗性)质粒混匀后按照P3细胞核转染试剂盒,使用程序EN150进行核转染。转染完成后将细胞平均分至30个10 cm培养皿中培养。24 h后将培养液换为含有浓度为1 mg/mL 的G418 培养基进行药物筛选,每3 d更换1次液体。
药筛至未转染阴性对照组细胞全部死亡时,将G418 浓度调整至500 μg/mL,继续培养7~10 d 至单克隆细胞形成。用克隆环挑取状态较好的单克隆细胞至24孔板中,待细胞长满后留取30%于原培养板提基因组,70%传代至6 孔板中至细胞长满后冻存备用。
使用细胞裂解液NP40裂解细胞提取基因组DNA并进行PCR 扩增,裂解反应条件为:55 ℃60 min,95 ℃10 min。PCR 产物测序后比对序列,选择突变的细胞基因组经PCR 扩增和琼脂糖凝胶纯化。将纯化的基因组DNA 与pMD18-T 载体连接后转化至大肠杆菌感受态中,每板挑选12 个菌落进行测序。测序结果与野生型比对后确定Sall1基因敲除细胞系。
2 结果
2.1 人、猪、鼠SALL1 氨基酸序列及蛋白结构比对结果
使用DNAMAN 进行氨基酸比对发现:人与猪SALL1氨基酸的相似度为92.46%,人与鼠SALL1氨基酸的相似度为90.19%。使用SOPMA在线网站进行二级结构比对发现:人SALL1 蛋白的α螺旋占比为23.79%,β 转角占比为4.53%,拓展链占比为13.44%,无规卷曲占比为58.23%;猪SALL1蛋白的α螺旋占比为23.23%,β转角占比为4.30%,拓展链占比为13.20%,无规卷曲占比为59.28%;鼠SALL1蛋白的α螺旋占比为22.98%,β转角占比为3.85%,拓展链占比为11.87%,无规卷曲占比为61.30%(图1)。使用PyMOL 软件进行SALL1 蛋白三维结构分析发现:人与猪三级结构对比时均方根偏差(root mean square deviation,RMSD)为0.155,人与鼠三级结构对比时RMSD 值为12.827(图2)。使用CD-search 进行结构域对比发现:人、猪SALL1 蛋白都具有SUF4-like、SUF4-like super family、zf-H2C2-2、zf-C2H2 4种结构域,而鼠SALL1蛋白具有SUF4-like、SUF4 -like super family、zf -H2C2 -2、zf -C2H2、COG5048这5种结构域(图3)。

图1 人、猪、鼠SALL1蛋白的二级结构构成Figure 1 Secondary structural composition of human,pig and mouse SALL1 protein
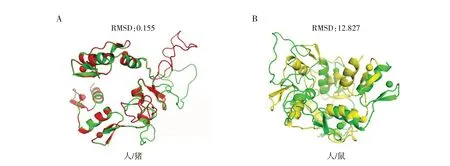
图2 人与猪、人与鼠SALL1蛋白三级结构比较Figure 2 Tertiary structure prediction of human/pig and human/mouse SALL1 protein

图3 人、猪、鼠SALL1蛋白结构域对比Figure 3 Comparison of human,pig and mouse SALL1 protein domains
2.2 CRISPR/Cas9敲除位点选择及打靶载体的构建
根据NCBI 中所提供的猪Sall1基因序列,在其两端设计扩增Sall1基因的PCR 引物(Sall1-F:5′-AACCGCACCACTAAGAGCAAGGAT-3′;Sall1-R:5′-AAGGGTTGGCAGATGTTCGTAAAG-3′),以猪PFF 基因组为模板,进行PCR 扩增,将其产物经琼脂糖凝胶电泳后得到Sall1基因的单一明亮电泳条带(图4),同时将其PCR 扩增产物送至生物公司测序,比对测序结果证明其序列与NCBI 中提供的序列一致。
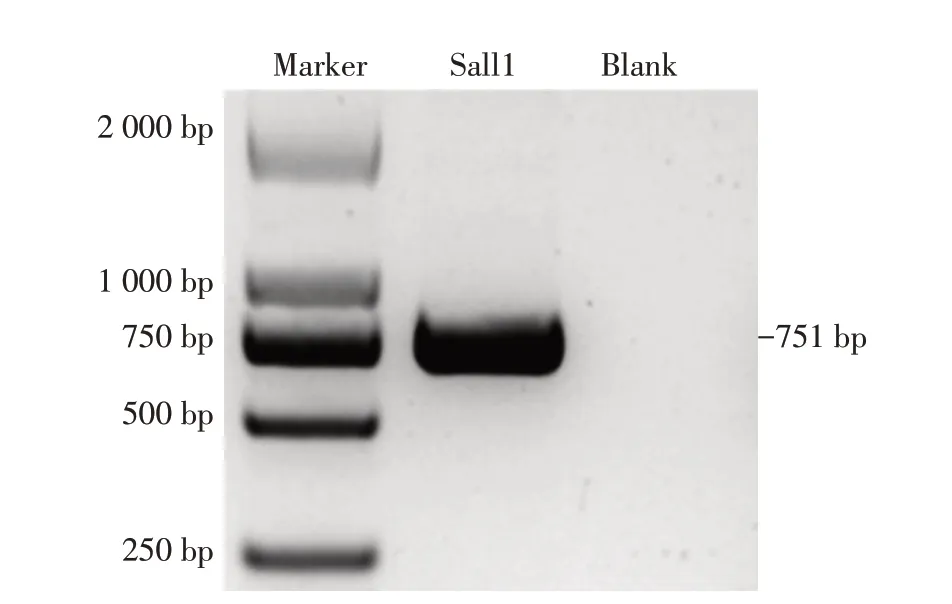
图4 Sall1基因PCR扩增产物凝胶电泳Figure 4 The results of PCR amplification products of Sall1
选取Sall1基因第1 外显子作为CRISPR/Cas9作用靶点设计sgRNA(图5),并在其两端加上可与BbsⅠ酶切位点相连接的黏性末端合成相应的Oligo(Sall1 -sgRNA -F:5′ -CACCGAACCTCTCTACCTCAACTCG-3′;Sall1-sgRNA-R:5′-AAACCGAGTTGAGGTAGAGAGGTTC-3′),将使用BbsⅠ酶切后的PX330质粒与退火后的Oligo连接,转化入大肠杆菌感受态后挑取单克隆菌落送至公司测序。测序结果证明PX330成功与sgRNA连接(图6)。
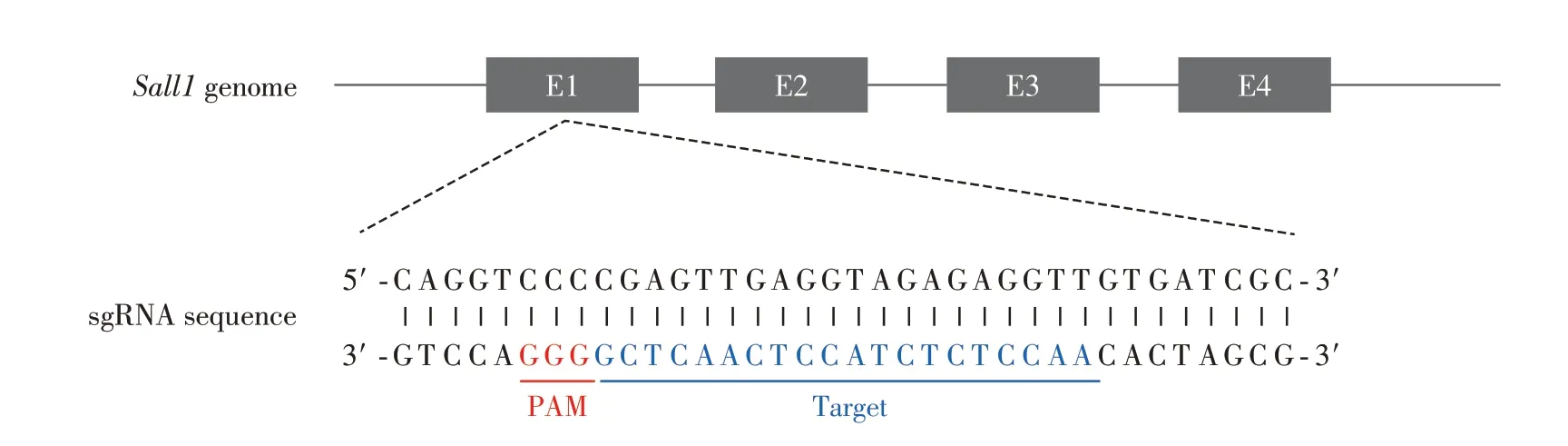
图5 Sall1基因sgRNA靶点设计模式图Figure 5 The pattern diagram of Sall1 gene knockout

图6 重组载体测序验证Figure 6 Verification of recombinant vector via sequencing
2.3 Sall1基因CRISPR/Cas9打靶载体效率验证
将重组打靶载体PX330-sgRNA 转染至PFF 后,提取细胞基因组。使用PCR 扩增其基因组后送至生物公司测序,测序结果表明PX330-sgRNA的打靶效率为51.6%。
2.4 Sall1基因敲除细胞系的制备及鉴定
将PX330-sgRNA 打靶载体与Td-tomato(G418抗性)质粒共同转染原代PFF 后,经G418 药筛获得33 个单克隆细胞系。TA 克隆测定具体序列,共获得23 个Sall1基因敲除的细胞系,敲除效率为69.7%。其中Sall1双等位基因敲除的细胞系16 个(主要敲除形式见表1),敲除效率为48.5%。

表1 Sall1-/-细胞系的主要敲除形式Table 1 The knocking-out modalities of Sall1-/-cell line
3 讨论
哺乳动物胚胎肾脏发生十分复杂,可分为前肾、中肾、后肾3个阶段。前肾和中肾在肾脏发生过程中逐渐退化,后肾最终发育为永久肾。胎儿发育晚期输尿管芽侵入后肾间质,随后这两种组织相互诱导共同分化促使后肾的发生。研究已发现许多对后肾发生至关重要的基因,主要包括Sall1、Six1、Gdnf、Six4、Eya1等。SALL1蛋白在S形体、逗号形小体、肾小管和足细胞等间充质衍生结构中大量表达。研究表明人类Sall1基因突变将会导致一种与多器官发育缺陷相关的常染色体显性遗传病,即Townes-Brocks 综合征,其可表现为肾发育不全或肾缺陷[21]。Sall1基因敲除的纯合子小鼠表现出输尿管芽生长及分支受损[22],这说明Sall1基因对后肾发育中输尿管芽的侵袭作用至关重要。
与啮齿类动物相比,猪的组织形态与生理和人更为相似[23]。同时,与非人灵长类动物相比,猪具有广泛的可用性、优良的育种潜力和较短的生殖成熟期[23]。本研究利用生物信息技术分析人、猪、鼠SALL1蛋白的氨基酸序列、二级结构、三级结构和保守结构域,发现猪SALL1蛋白与人SALL1蛋白具有高度同源性。由此本文认为,制备Sall1基因敲除的猪PFF细胞系,以获得肾脏缺失的猪模型,使下一步通过猪Sall1基因敲除囊胚和人干细胞互补在猪体内再生人源化肾脏成为可能。
早期传统的基因敲除方法基于同源重组,其用途也主要局限于小鼠模型的开发[24]。2013 年CRISPR/Cas9基因编辑系统的问世极大地促进了大动物基因编辑模型的发展。本课题组在前期研究中已成功构建多种基因敲除的猪模型,如C3-/-、Six1-/-和Six1-/-/Six4-/-等基因敲除克隆猪[12,25],这验证了CRISPR/Cas9在大动物基因编辑模型构建中的可行性与便捷性。本研究首先通过测序检测用于基因编辑的Sall1-PX330-sgRNA 打靶载体的敲除效率,随后将其与G418抗性的质粒共同转染入猪PFF细胞系中。通过药物筛选共获得33 个单克隆细胞系,经过多重鉴定后发现共有23 个Sall1单等位基因和双等位基因敲除的细胞系,其敲除效率高达69.7%,而其中双等位基因敲除的细胞系有16个,其敲除效率为48.5%。本研究结果显示利用CRISPR/Cas9 技术制备Sall1基因敲除的猪PFF 细胞系具有高效性与可实施性。
综上所述,本研究利用生物信息学方法验证了人与猪SALL1 蛋白较高的同源性。同时构建了可用于Sall1基因敲除的CRISPR/Cas9打靶载体,并成功获得了Sall1双等位基因敲除的细胞系,这些细胞系可为构建猪肾脏缺失模型提供供体细胞,为探究Sall1基因在猪肾脏发育过程中的作用提供前期基础,并为在异种动物体内再生人源化肾脏提供理论依据。
猜你喜欢
——一道江苏高考题的奥秘解读和拓展
中学生物学(2022年7期)2022-09-07
成都医学院学报(2022年4期)2022-08-19
河北医科大学学报(2022年2期)2022-03-15
科学与生活(2021年16期)2021-11-25
宁夏医学杂志(2021年7期)2021-08-08
江西农业学报(2021年4期)2021-04-20
安徽医学(2020年11期)2020-12-25
实用医学杂志(2020年16期)2020-09-25
三农资讯半月报(2020年11期)2020-06-21
医学研究杂志(2015年9期)2015-07-01
