
ALCAT1缺失在KRAS诱导肺腺癌中的作用及机制研究
2022-07-16 04:59张潇月史裕光
南京医科大学学报(自然科学版) 2022年7期
张潇月,郑 月,蒋 厅,史裕光
南京医科大学生物化学与分子生物学系,江苏 南京 211166
目前在全球范围内,肺癌仍然是导致癌症死亡的主要原因[1]。肺癌患者中约80%为非小细胞肺癌,其中肺腺癌是非小细胞肺癌最常见的亚型[2]。在肺腺癌中Kirsten 大鼠肉瘤病毒癌基因同源物(Kirsten rat sarcoma viral oncogene homolog,KRAS)第12 号密码子经常发生突变[3]。突变的KRAS 通过调节Raf/Erk 和PI3K/Akt 信号通路促进肺腺癌的发生[4],并导致高度活跃的糖酵解和氧化应激增加[5]。到目前为止,KRAS 突变的肺腺癌没有有效的治疗方法。
肿瘤缺氧微环境导致癌细胞代谢异常和快速增殖[6]。肿瘤代谢异常会激活PI3K/Akt信号通路,该通路通过关键转录因子缺氧诱导因子1α(hypoxiainducible factor 1 alpha,HIF1α)调节糖酵解,并降低线粒体功能[7-8]。线粒体功能障碍会导致活性氧(reactive oxygen species,ROS)增加,研究发现增加ROS水平可促进癌细胞转移[9]。此外肿瘤中ROS增加可以激活PI3K/Akt 通路调节HIF1α的转录,从而促进氧化磷酸化到糖酵解的转变[10],并且ROS对KRAS 诱导肺腺癌的发生发展至关重要[11],但ROS 在KRAS 诱导的肺腺癌中的调节机制尚未完全清楚。
心磷脂(cardilipin,CL)是一种线粒体标志性磷脂,主要存在于线粒体内膜中,在线粒体功能中发挥重要作用,包括电子传递、线粒体动力学、线粒体生物发生、凋亡机制和血管发育[12]。CL 酰基链结构决定其功能,在正常组织中以亚油酸(C18∶2)为主。衰老引起心磷脂的病态重构,导致亚油酸(C18∶2)逐渐被二十二碳六烯酸(docosahexaenoic acid,DHA,C22∶6)所取代,DHA 富含双键使得CL对ROS 的氧化损伤高度敏感,氧化的CL 导致线粒体功能障碍和各种衰老相关疾病[13]。研究发现在C6 神经胶质瘤细胞中CL 酰基链改变导致能量代谢从氧化磷酸化转变为糖酵解[14]。此外CL分子种类异常与胶质瘤细胞和甲状腺癌细胞增殖相关[15-16]。
心磷脂酰基转移酶1(cardiolipin acyltransferase 1,ALCAT1)是一种酰基转移酶,通过催化溶血CL再酰化为CL,导致CL 酰基链DHA 含量增加[17]。本实验室前期工作发现在各种衰老相关代谢疾病中ALCAT1 高表达并导致线粒体功能障碍。ALCAT1基因缺失通过阻止ROS产生恢复线粒体呼吸,并预防各种与年龄相关疾病的发生,包括神经退行性疾病[18]和心血管疾病[19]等。研究发现ALCAT1在人类非小细胞肺癌中显著上调,利用ALCAT1 模拟肽阻断ALCAT1 活性可影响非小细胞肺癌的迁移[20]。本研究通过构建CC10rtta-KRAS/ALCAT1 KO 转基因小鼠模型,初步发现ALCAT1 基因缺失减缓KRAS 诱导肺腺癌的发展,并发现在A549细胞中抑制ALCAT1 可阻止ROS 产生,降低HIF1α表达,从而抑制糖酵解代谢产物,并影响Akt 磷酸化水平,为进一步研究ALCAT1 缺失或抑制在肺腺癌中的作用与机制奠定了基础。
1 材料和方法
1.1 材料
人肺腺癌细胞系(A549)购自中国科学院生物化学与细胞生物学研究所;CC10rtta-KRAS小鼠来自清华大学,ALCAT1 KO 小鼠由本实验室前期构建[17],在南京医科大学实验动物基地的SPF 环境中饲养,研究经南京医科大学实验动物福利伦理委员会批准,批文编号:IACUC-1601272-1。4%多聚甲醛(武汉塞维尔生物公司),GAPDH、HIF1α、p-Akt(T308)、Akt、Erk1/2、p-Erk1/2 抗体(Cell Signaling Technology公司,美国),ECL 显影液(Thermo Fisher 公司,美国),HiScriptⅡQ RT SuperMix、ChamQ SYBR qPCR Master Mix(南京诺维赞公司),引物由上海杰瑞公司合成,HE 染色试剂(北京索莱宝科技公司),ROS 检测试剂盒(上海碧云天科技公司),乳酸检测试剂盒(南京建成生物公司),多西环素饲料(北京凯国科技有限公司),Jenuglitapin(Jenu,Perenna Pharmaceuticals公司,美国),Mito Q(MedChem Express公司,美国)。
1.2 方法
1.2.1 动物分组和处理
将ALCAT1 KO 小鼠与CC10rtta-KRAS 小鼠杂交,最终产生CC10rtta-KRAS/ALCAT1 KO小鼠,对照组为CC10rtta-KRAS 小鼠。8 周龄CC10rtta-KRAS/ALCAT1 KO、CC10rtta-KRAS 雄性小鼠随机分为正常饮食组(n=3)及多西环素饮食组(n=6)。分别在多西环素饮食8 周、12 周通过小动物CT 观察肺部肿瘤,在实验终点多西环素饮食12 周时解剖取肺组织。
1.2.2 基因型鉴定
从小鼠脚趾中提取DNA,利用Rapid Taq master mix、引物、双蒸水配制PCR 体系,进行目的片段扩增。根据电泳结果,确定其基因型以筛选出符合要求的小鼠。PCR 扩增引物序列:NEO 正向引物5′-AGGATCTCCTGTCATTCTACCTTGCTC-3′,反向引物5′-AAGAACTCGTCAAGAAGGCGATAGAAGGCG-3′;ALCAT1正向引物5′-GCCCTCATCAGCACTCAGTA-3′,反向引物5′-AAAACACAAAATGGATATGCAGAA-3′;CC10rtta 正向引物5′-AAAATCTTGCCAGCTTTCCCC-3′,反向引 物5′-ACTGCCCATTGCCCAAACAC-3′;KRAS 正向引 物5′-AGACACAAAACAGGCTCAGGA-3′,反向引物5′-GGAGACAATGGTTGTCAACAGA-3′。
1.2.3 微计算机断层扫描技术
使用微计算机断层扫描技术(micro-computed tomography,micro-CT)观察小鼠肺部肿瘤。正式扫描标本前,先扫描标准模体,进行CT 值校正。使用2%异氟烷,98%氧气条件下对小鼠进行麻醉,选择胸腔视野进行扫描,调节参数。每只小鼠扫描时间约20 min。扫描完成后,使用IRW(Inveon research workplace)软件进行图像分析。
1.2.4 Western blot检测
将组织或收集的细胞裂解,离心取上清。使用BCA 蛋白浓度检测试剂盒进行蛋白定量。将上样缓冲液与蛋白样品混匀,95 ℃加热10 min,蛋白充分变性后置于冰上,并逐一上样。将20 μg 蛋白质在10%或15%SDS-聚丙烯酰胺凝胶上进行电泳,调节电压至80 V,待样品进入分离胶后,换成120 V继续;电泳结束后,转移到PVDF膜上,PVDF膜用甲醇激活后,按顺序放在转膜夹中,在4 ℃、90 V、2 h 的条件下转膜。用5%脱脂牛奶室温封闭1 h,用5%BSA 配制一抗(1∶1 000),4 ℃孵育过夜,回收抗体,洗膜,在室温下与二抗孵育1 h,回收二抗,洗膜。ECL发光液均匀滴加在PVDF膜上,曝光。
1.2.5 HE染色
小鼠麻醉后颈椎脱臼法牺牲小鼠,打开胸腔取出肺脏,随后将肺组织固定在4%多聚甲醛中,48 h后进行修块、脱水、包埋,制好切片标本。切片在二甲苯中脱蜡并使用梯度乙醇再水合,蒸馏水洗3 min,苏木素染细胞核1 min,水洗5 min,酒精盐酸分色(1%的盐酸加入70%乙醇中)数秒,快速放入水中,再用流水冲洗蓝化5 min,伊红染细胞质1~2 min,复水透明,中性树脂封片,镜下拍照。
1.2.6 RT-PCR
肺组织加入TRIzol 匀浆,离心取上清,提取RNA。使用HiScript ⅡQ RT SuperMix 将提取的RNA逆转录为cDNA,将cDNA稀释至100 ng/μL,使用ChamQ SYBR qPCR Master Mix 进行RT-PCR扩增检测基因表达,PCR 程序如下:95 ℃预变性5 min,95 ℃变性30 s,60 ℃退火延伸60 s,95 ℃1 min,循环40 次。GAPDH 用作mRNA 内部标准化对照。RT-PCR 扩增引物包括以下序列:TTF1 正向引物5′-AAAACTGCGGGGATCTGAG-3′,反向引物5′-TGCTTTGGACTCATCGACAT-3′;GAPDH 正向引物5′-AATGGTGAAGGTCGGTGTG-3′,反向引物5′-GTGGAGTCATACTGGAACATGTAG-3′。
1.2.7 细胞培养
A549 细胞使用DMEM 高糖培养基培养,并在培养基中加入10%胎牛血清(FBS)、100 U/mL 青霉素和100 mg/mL 链霉素,放在37 ℃、5%CO2培养箱中培养。
1.2.8 乳酸检测
将A549 细胞接种到6 孔板,分为4 组:对照组、Jenu 组、缺氧组、缺氧+Jenu 组,CoCl2200 μmol/L 诱导缺氧,Jenu 10 μmol/L 抑制ALCAT1,共处理细胞24 h,收集细胞培养的上清液,使用乳酸检测试剂盒检测上清液中乳酸的量。
1.2.9 ROS检测
将A549细胞接种到6孔板,首先阳性对照组加入ROS up处理30 min,随后所有细胞均加入DCFH-DA荧光探针孵育,30 min 后弃培养液,用PBS 洗两遍,细胞重悬于0.5 mL PBS 并转移到EP 管中,取100 μL加入到酶标板中,用荧光酶标仪检测。剩余的细胞悬液裂解后测蛋白浓度,最后ROS用蛋白浓度校准。
1.3 统计学方法
数据均使用Graphpad Prism 6 软件进行统计分析,并作图。所示数据以均数±标准差()表示。组间比较采用t检验,P<0.05 为差异有统计学意义。
2 结果
2.1 CC10rtta-KRAS/ALCAT1 KO 小鼠肺癌模型的构建
四环素操纵子(TetO)调节的KRAS(TetO-KRAS)小鼠与Clara细胞分泌蛋白(CC10)反向四环素反式激活因子(rtta)即CC10rtta 小鼠杂交,当多西环素饮食时,KRAS-G12D 位点被激活,在呼吸道上皮细胞特异性表达诱导肺腺癌模型[21]。为了验证ALCAT1 基因的缺失是否可以缓解KRAS 诱导的肺腺癌的发生,本研究将ALCAT1 基因表达缺失的小鼠(ALCAT1 KO)与CC10rtta-KRAS 小鼠杂交,最终得到实验组CC10rtta-KRAS/ALCAT KO(以下简称CK/ALCAT1 KO)转基因小鼠,对照组CC10rtta-KRAS(以下简称CK)小鼠(图1A)。同时对各组小鼠进行基因型鉴定,对于CK小鼠,检测的是敲入的CC10rtta-KRAS 基因,当小鼠表达该基因时,PCR 可以检测出CC10rtta-KRAS 条带,反之该基因未导入到小鼠体内;对于ALCAT1 KO 小鼠,引物ALCAT1检测小鼠是否表达ALCAT1 基因,引物NEO 检测小鼠ALCAT1 基因是否被成功敲除。结果证实CC10rtta-KRAS 基因双转入并且ALCAT1 敲除,CC10rtta-KRAS/ALCAT1 KO 小鼠模型构建成功(图1B)。本研究选取8周龄的小鼠,分别在多西环素饮食8周、12周时,通过micro-CT检测小鼠肺部的肿瘤发生情况,并在12周实验终点解剖取小鼠肺组织观察肺腺癌的发展(图1C)。从小鼠8 周龄开始记录体重,即多西环素饮食诱导的0~12 周,发现正常饮食组体重无显著差异,CK小鼠从多西环素饮食第5周开始,体重增长显著减慢(图1D)。
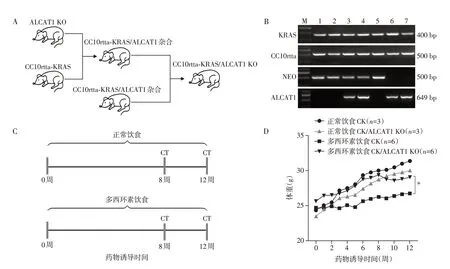
图1 CC10rtta-KRAS/ALCAT1 KO小鼠肺癌模型的构建Figure 1 Constructed lung cancer model of CC10rtta-KRAS/ALCAT1 KO mice
2.2 micro-CT检测小鼠的肺部肿瘤
多西环素饮食驱动KRAS 的表达诱导肺腺癌,在多西环素饮食8 周时通过micro-CT 扫描小鼠胸部,结果显示,多西环素饮食组CK/ALCAT1 KO小鼠的肺部未检测到肿瘤,在CK小鼠中检测到两只小鼠肺部有肿瘤,其中一只右半肺被肿瘤侵蚀,正常饮食组未发生肿瘤(图2A)。为了进一步研究ALCAT1缺失在肺腺癌中的作用,将多西环素饮食诱导时间持续到12 周,结果显示与多西环素饮食8 周相比,增加多西环素饮食时间加剧肺部肿瘤发展。多西环素饮食12周时,CK组中观察到100%肺部肿瘤的发生,但肿瘤体积存在差异;CK/ALCAT1 KO 小鼠的肺部肿瘤体积相对较小,且部分小鼠未观察到肿瘤(图2B),提示ALCAT1基因缺失影响肺部肿瘤的发展。
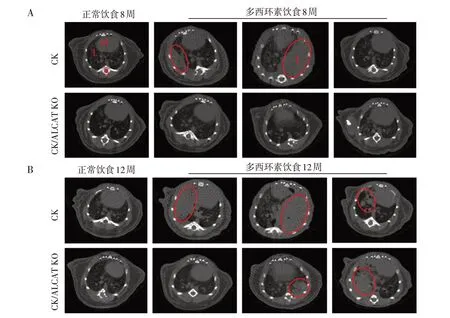
图2 micro-CT检测小鼠的肺部肿瘤Figure 2 micro-CT detected lung tumors in mice
2.3 ALCAT1缺失减缓KRAS诱导的肺腺癌的发展
将多西环素饮食的时间延长到12 周加剧肺部肿瘤的形成,并在实验终点将小鼠解剖取肺组织,结果显示正常饮食组无肿瘤发生;多西环素饮食组中与CK/ALCAT1 KO小鼠相比,CK小鼠肺部肿瘤对肺的侵蚀程度更高(图3A)。对肺重/体重进行相对定量分析,发现多西环素饮食组中CK 小鼠肺重/体重值明显高于CK/ALCAT1 KO 小鼠和正常饮食组小鼠(图3B)。TTF1 是肺腺癌标志物,RT-PCR 检测结果显示,与正常饮食组相比,多西环素饮食组TTF1 的表达明显升高,提示肺腺癌的存在,而ALCAT1 缺失降低TTF1 的表达(图3C)。HE 染色结果显示,正常饮食组的小鼠肺部可见结构清晰的肺泡,多西环素饮食组中CK 小鼠出现多个大的固体型腺癌灶,肺部结构被侵占,CK/ALCAT1 KO小鼠减少局灶性增生,癌灶体积缩小(图3D)。以上结果提示ALCAT1的缺失减缓KRAS 诱导的肺腺癌的发展。

图3 ALCAT1缺失减缓KRAS诱导的肺腺癌的发展Figure 3 ALCAT1 deficiency prevented the development of KRAS induced lung adenocarcinoma
2.4 ALCAT1缺失降低Akt磷酸化水平
为了进一步研究ALCAT1 缺失是否通过影响KRAS 基因相关通路延缓肺腺癌的发展,本研究通过Western blot检测Akt、Erk1/2蛋白的磷酸化水平,结果显示在多西环素饮食组中,与CK 小鼠相比,ALCAT1 缺失降低Akt 磷酸化水平(图4A),灰度分析发现CK/ALCAT1 KO 小鼠中p-Akt(T308)/Akt 比值下降(图4B),但CK 小鼠与CK/ALCAT1 KO 小鼠之间p-Erk1/2/Erk1/2比值差异不明显(图4C)。
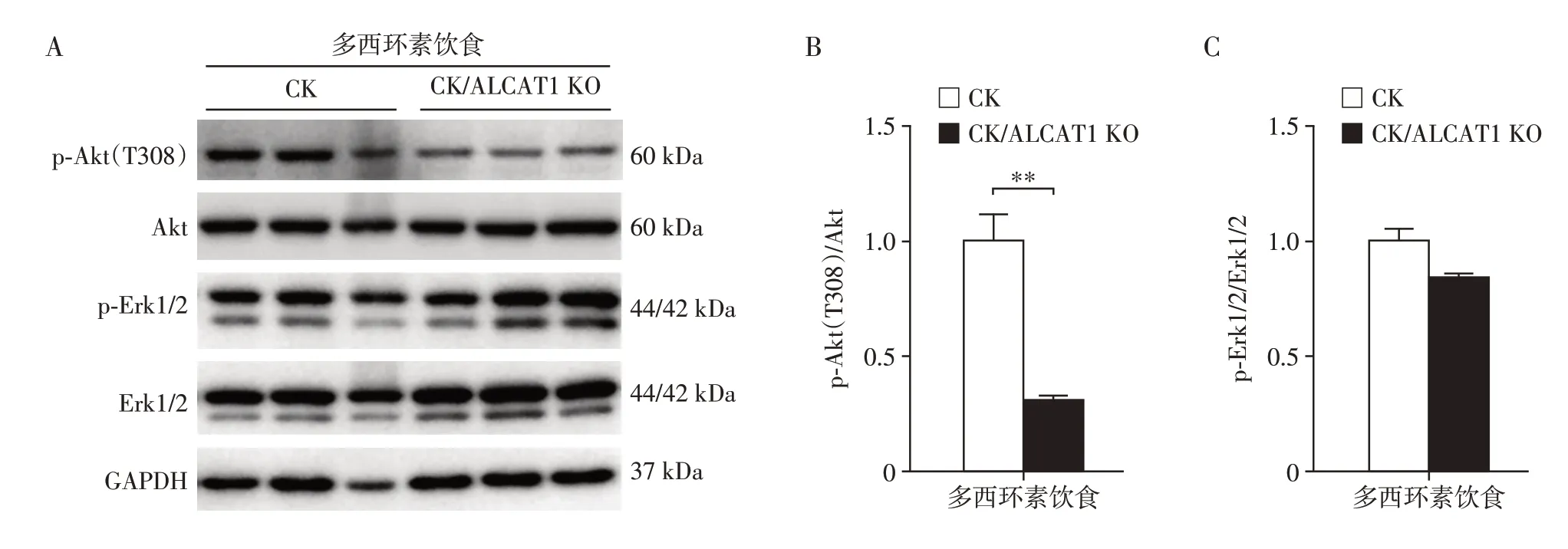
图4 ALCAT1的缺失降低Akt磷酸化水平Figure 4 ALCAT1 deficiency reduced Akt phosphorylation levels
2.5 抑制ALCAT1通过阻止ROS产生抑制HIF1α表达,减少糖酵解代谢,影响Akt磷酸化水平
为了进一步研究ALCAT1 在KRAS 突变的肺腺癌中的作用机制,本研究利用CoCl2(200 μmol/L)处理KRAS突变的人肺腺癌细胞系(A549)诱导缺氧微环境激活HIF1α,并联合ALCAT1 抑制剂(Jenu)药理学抑制ALCAT1。通过Western blot 检测Akt 磷酸化水平,结果显示,在缺氧条件下Akt磷酸化水平明显升高,Akt磷酸化水平随Jenu浓度增加表达下降,Jenu(10 μmol/L)抑制效果显著(图5A、B)。与未缺氧处理的对照组相比,缺氧处理后ROS 水平增加,Jenu 抑制ALCAT1 在缺氧/未缺氧条件下都显著降低A549 细胞中的ROS 水平(图5C)。此外与对照组相比,缺氧增加糖酵解产物乳酸生成,Jenu 抑制ALCAT1 显著降低乳酸的生成量(图5D)。在A549细胞中使用线粒体抗氧化剂(Mito Q)抑制线粒体ROS 的产生,Western blot 检测结果显示缺氧诱导HIF1α激活,Mito Q(5 μmol/L)抑制ROS的产生,显著降低HIF1α的表达,同时发现Jenu(3 μmol/L)处理条件下HIF1α的表达降低不明显,Jenu(10 μmol/L)显著降低HIF1α的表达(图5E、F)。上述结果证明在缺氧条件下抑制ALCAT1通过阻止ROS产生抑制HIF1α表达,从而减少糖酵解代谢,并影响Akt 磷酸化水平。
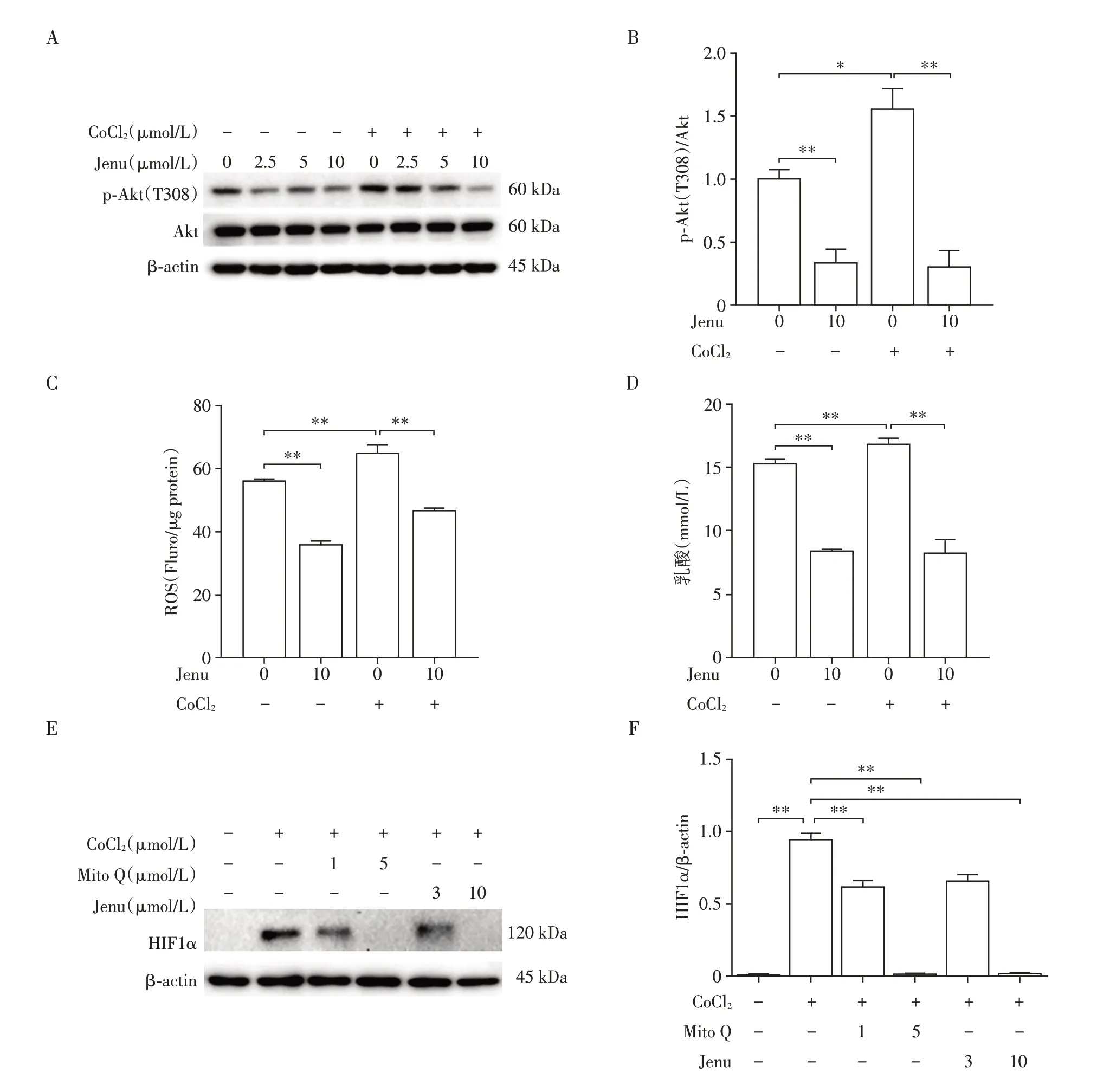
图5 抑制ALCAT1通过阻止ROS产生抑制HIF1α表达,减少糖酵解代谢,影响Akt磷酸化水平Figure 5 Inhibition of ALCAT1 attenuated HIF1α expression by preventing ROS production,reduced glycolytic metabolism,affected Akt phosphorylation levels
3 讨论
越来越多证据表明癌症是衰老相关疾病,癌症发病率随年龄的增加而升高,癌症与衰老的分子学机制都包括线粒体DNA(mtDNA)突变、能量代谢异常和ROS水平增加[9]。衰老显著增加DHA在CL酰基链中的含量和CL 脂质过氧化,氧化的CL 进一步导致线粒体功能障碍和衰老相关疾病的发生[13]。目前已经发现CL 分子种类异常与肿瘤细胞增殖相关[16]。ALCAT1催化溶血心磷脂再酰化为CL,导致CL酰基链DHA含量增加并对ROS氧化损伤更加敏感。在各种衰老相关代谢疾病中发现ALCAT1高表达并导致线粒体功能障碍和ROS 水平增加[18]。ALCAT1基因缺失通过阻止ROS产生减弱HIF1α表达并缓解线粒体功能障碍,预防各种与年龄相关疾病的发生[19]。此外,ALCAT1 在血管生成中发挥了十分重要的作用,ALCAT1 siRNA 降低了内皮基因(VEGFR2 和CD31)的表达。最近研究发现利用ALCAT1 模拟肽阻断ALCAT1 活性影响非小细胞肺癌的迁移[20]。本研究初步发现抑制ALCAT1通过降低ROS 水平下调HIF1α的表达,降低糖酵解代谢产物生成,从而影响KRAS诱导的肺腺癌的发展。
约25%的肺腺癌是KRAS 功能获得性突变导致的,但目前针对KRAS 突变的肺腺癌没有有效治疗方法[22]。CC10rtta-KRAS 条件性敲入小鼠模型被广泛用于肺腺癌的研究,小鼠经多西环素诱导后,KRAS 在呼吸道上皮特异性表达诱发肺腺癌[23]。本研究将CC10rtta-KRAS 小鼠与ALCAT1 KO 小鼠杂交构建CC10rtta-KRAS/ALCAT1 KO 小鼠肺腺癌模型,通过PCR 验证CC10rtta-KRAS 基因的敲入和ALCAT1 基因敲除,为进一步研究ALCAT1 缺失对KRAS 肺腺癌的影响提供模型。由于KRAS 基因缺乏可成药的位点,很难开发靶向治疗药物[24]。最近研究发现,KRAS 突变的肺癌高度依赖糖酵解代谢,这种代谢依赖性提供了靶向治疗的机会[25]。
当多西环素饮食启动KRAS表达后,通过micro-CT在小鼠肺部检测到肿瘤,与猜想一致的是相比于CK小鼠,ALCAT1缺失延缓肿瘤的发生,并对肺的侵蚀程度降低。同时发现小鼠肺部肿瘤体积随多西环素饮食时间增长而增大。根据恶性肿瘤的细胞学特征[26],通过HE 切片观察发现小鼠肺部肿瘤具备肺腺癌的特征,而ALCAT1 缺失减少肿瘤局灶性增生,减缓肺腺癌的发展。在肺腺癌中KRAS 突变可以激活Raf/Erk 和PI3K/Akt 信号通路促进肿瘤发生发展。Raf/Erk 或PI3K/Akt 是糖酵解代谢中重要的信号转导途径,直接或间接上调糖酵解[4]。本研究发现在KRAS 诱导的肺腺癌中ALCAT1 缺失降低Akt磷酸化水平,但ALCAT1缺失对Erk磷酸化水平影响不大。
肿瘤细胞为了适应低氧环境会通过减少耗氧量增加糖酵解代谢或激活一些基因的表达恢复供需平衡、促进肿瘤生长。HIF1α转录因子在大多肿瘤中是激活的,其维持糖酵解并且调节线粒体呼吸,激活的HIF1α还可调节细胞增殖、迁移和血管生成的基因[27]。研究发现在肿瘤中ROS 水平增加并激活代谢相关通路,如PI3K/Akt 诱导HIF1α转录活性[10]。本研究使用CoCl2处理A549 细胞诱导缺氧,激活HIF1α,模拟实体肿瘤的缺氧微环境,不同浓度的Jenu抑制ALCAT1表达,发现A549细胞在缺氧的条件下,ROS的产生水平增加,ALCAT1抑制显著降低缺氧条件下ROS 的产生。并发现在缺氧条件下抑制ALCAT1减少糖酵解产物乳酸生成,改善A549细胞异常的代谢。为了进一步研究ROS在KRAS诱导的肺腺癌中的调节机制,本研究使用Mito Q 有效清除线粒体ROS的产生,可以显著降低HIF1α表达,同时发现抑制ALCAT1 也显著降低HIF1α表达,提示ALCAT1 抑制剂与抗氧化剂有同样抑制效果,通过抑制ROS 水平降低HIF1α表达。本研究发现在A549细胞中缺氧导致Akt 磷酸化水平增加,而抑制ALCAT1显著降低Akt磷酸化,但并未发现抑制ROS产生直接影响PI3K/Akt 通路降低HIF1α表达,未来将进一步研究ALCAT1-ROS-HIF1α轴背后的分子事件以及与线粒体呼吸和线粒体功能联系,并将在更多的癌症模型中去验证ALCAT1 缺失对肿瘤发生发展的影响。
综上所述,ALCAT1 的缺失减缓KRAS 突变导致的肺腺癌的发展,在KRAS 突变的人肺腺癌细胞中药理学抑制ALCAT1 可能通过阻止ROS 的产生降低HIF1α表达,从而减少糖酵解代谢产物的生成,影响Akt 磷酸化水平。这为开发新型抗肺癌药物提供了新的干预靶点,也为后续机制研究提供了基础。
猜你喜欢
华夏医学(2021年1期)2021-12-05
实用老年医学(2021年7期)2021-12-04
皮肤病与性病(2021年3期)2021-07-30
当代水产(2021年3期)2021-07-20
视界观·上半月(2020年2期)2020-04-20
读与写·上旬刊(2018年1期)2018-05-06
美与时代·城市版(2018年10期)2018-01-16
教育教学论坛(2016年51期)2017-03-22
教育教学论坛(2016年49期)2017-02-27
癌变·畸变·突变(2015年3期)2015-02-27
