
口服抗凝剂治疗以新生儿暴发性紫癜或颅内出血为首发表现的遗传性复合杂合突变的重度蛋白C缺乏症2例病例报告并文献复习
2022-06-14 03:20宋予晴唐晓艳全美盈孙之星李蕴微马明圣赵永强
中国循证儿科杂志 2022年1期
宋予晴 肖 娟 唐晓艳 李 卓 全美盈 孙之星 李蕴微 马明圣 赵永强
遗传性蛋白C缺乏症(PCD)是一种常染色体显性遗传性罕见病,主要表现为蛋白C水平降低,其所致血栓占血栓栓塞事件中的3%~5%[1],发病率1/40 000~1/250 000[1-3]。编码蛋白C的PROC基因位于染色体2q13-q14,由9个外显子组成,长约11.2 kb[4]。遗传性PCD患儿多为单碱基的杂合基因突变,临床表现为无症状或轻微症状,以下肢静脉血栓多见。纯合或复合杂合突变患儿更罕见,表现为蛋白C水平明显降低,部分患儿甚至<1%,多表现为新生儿期起病的危及生命的严重暴发性紫癜(PF)等[5],由于我国尚无浓缩蛋白C制剂的生产和供应,只能使用新鲜冰冻血浆(FFP)进行暂时救治,但几乎所有病例终因家长不能坚持长期治疗而死亡。本文报告2例中国医学科学院北京协和医院(我院)儿科口服抗凝剂成功救治以新生儿PF或颅内出血为首发表现的遗传性复合杂合突变的重度PCD并随访3和6年至今存活的患儿,希望帮助儿科医生(尤其是新生儿医生)加强对重度遗传性PCD的认识,早期诊断,积极治疗,改善预后。
1 病例资料
例1,女,6月龄,因“生后皮肤瘀斑反复发作”入我院。足月剖宫产分娩,出生体重2 800 g,生后1、5、10 min的Apgar评分均为10分。生后18 h起全身多处逐渐出现大面积紫癜及红斑样皮疹,中央破溃,可见黄绿色液体渗出(图1)。
就诊当地医院查血常规、CRP、肝肾功能正常,血培养阴性,破溃处创面培养出肺炎克雷伯菌。诊断为新生儿感染、败血症。予多种抗生素治疗无效。4月龄就诊上级医院,查PT/INR、APTT延长,纤维蛋白原(Fbg)降低,D-二聚体升高。考虑诊断先天性低纤维蛋白原血症,予FFP和冷沉淀治疗后紫癜可消退,停用FFP后再次出现新发瘀斑。生后6月来我院就诊,Fbg降低,D-二聚体升高(表1)。行易栓因素筛查:蛋白C显著降低,蛋白S、抗凝血酶Ⅲ及活化蛋白C(APC)抵抗正常范围。患儿姐姐生后出现皮肤紫癜、颅内出血,生后4 d死亡。结合患儿临床表现、蛋白C水平及家族史,拟诊断PCD。进一步行PROC基因突变分析,患儿为复合杂合突变,分别来源于父母,患儿父母的蛋白C水平均低于正常值,明确诊断遗传性复合杂合突变重度PCD。

表1 2例遗传性复合杂合突变重度PCD患儿主诉至随访临床事件
予FFP输注,联用低分子肝素(LMWH)抗凝治疗,患儿皮损逐渐修复,且无新发血栓形成,Fbg和D-二聚体恢复至正常范围(表1)。LMWH应用5 d后,加维生素K拮抗剂(VKA)华法林,监测INR达2.5~3.5时,逐渐减停FFP,单用华法林维持目标INR。随访至6岁,持续口服华法林,INR 2.63~3.41(表1),无新发血栓形成,生长发育正常,目前读小学一年级。
例2,男,生后6月因“颅内出血、肺出血,反复皮肤瘀斑”入我院。足月剖宫产分娩,出生体重3 150 g,生后1、5、10 min的Apgar评分均为10分。生后7 h因精神反应弱就诊当地医院,无皮肤瘀斑或紫癜样皮疹,血常规大致在正常范围,APTT正常,Fbg轻度降低,D-二聚体轻度升高(表1),纤维蛋白/纤维蛋白原降解产物(FDP)升高,头颅CT示多发颅内出血(图2A),肺部CT示双侧肺出血(图2B)。当地医院予冷沉淀纠正凝血、支持治疗等,1月龄复查头颅MR示出血灶较前好转。2月龄起,患儿反复出现皮肤瘀斑,且出现频率逐渐增加,伴多发血肿形成(表1)。6月龄来我院查PT、APTT稍延长,Fbg降低,D-二聚体、FDP显著升高(表1),易栓因素筛查:蛋白C显著降低,蛋白S、抗凝血酶Ⅲ及APC抵抗正常范围。诊断重度PCD,DIC,纤溶亢进。行PROC基因突变检测,患儿为复合杂合突变,分别来源于父母,患儿父母蛋白C水平均略有降低,明确诊断遗传性复合杂合突变重度PCD。予FFP输注,LMWH抗凝,患儿皮肤瘀斑改善,Fbg升至正常范围、D-二聚体和FDP均较前明显下降(表1)。基于文献证据,在我院医务处备案,且经患儿家长知情同意签字后,予LMWH应用3周后,调整为口服抗凝剂利伐沙班,初始剂量2.8 mg,q8h(基于EINSTEIN-Jr Ⅲ期[6]研究中采用的根据体重调整的利伐沙班给药方案),逐渐降低FFP输注频率至每周1次。随访至3.5岁,除新生儿脑出血导致的轻微的下肢运动障碍及语言障碍外,余生长发育均与同龄儿相仿。末次随访口服利伐沙班减量至2.4 mg,q8h,仅于新发皮肤瘀斑时予FFP间断输注治疗,未观察到利伐沙班不良反应。
2 文献复习
1991年首次报道用蛋白C制剂治疗PCD,在发达国家现已经成为主要治疗方法。我国一直没有浓缩蛋白C制剂的生产和供应,国内PCD主要治疗是输血浆,平均每周1次,病毒感染的风险大,一般家庭难以长期坚持。鉴于本文目的是回顾中国婴幼儿PCD诊断治疗现况,故以“protein C deficiency”及“Chinese”为关键词在PubMed数据库检索,以“蛋白C缺乏”为关键词在中国知网及万方数据库检索,从建库检索至2021年10月31日,逐篇阅读筛选婴儿期起病的遗传性PCD的中国病例。共检索到11篇(11例)目标文献,4例发表在英文期刊,余发表在中文期刊。
表2显示文献报道的11例遗传性PCD的临床资料[7-17],均为病例报告,最早报告于1998年,首次通过病理和临床诊断PCD,其余均在2011年后报告。合并本文2例共13例。12例于新生儿期起病,其中10例于生后48 h内即出现临床表现,最早于生后4 h发病,例6生后4月起病。除本文例2外,余12例均以典型的PF起病,即血栓引起的紫癜样皮损,其中例8合并左侧肾梗死,例4生后16 d头颅MR提示脑血栓伴颅内出血,例3和9则分别于生后8和16 d头颅影像学提示颅内出血。例5 FDP>150(正常值<5)mg·L-1,可能存在DIC。例2~12蛋白C水平0~8%,均显著降低,9例通过基因检测确诊为遗传性PCD,其中2例为PROC基因纯合突变,7例为复合杂合突变。在例1~11中,例7未报道治疗及结局,例6每日输注FFP 15 mL·kg-1×40 d获得临床缓解,后隔日输注FFP共3月,随访至13月龄仍存活,紫癜样皮损好转且监测无新发血栓形成,7例单用FFP或联合LMWH曾获短暂临床缓解,6例因家长选择放弃治疗,本文2例治疗情况见前文。
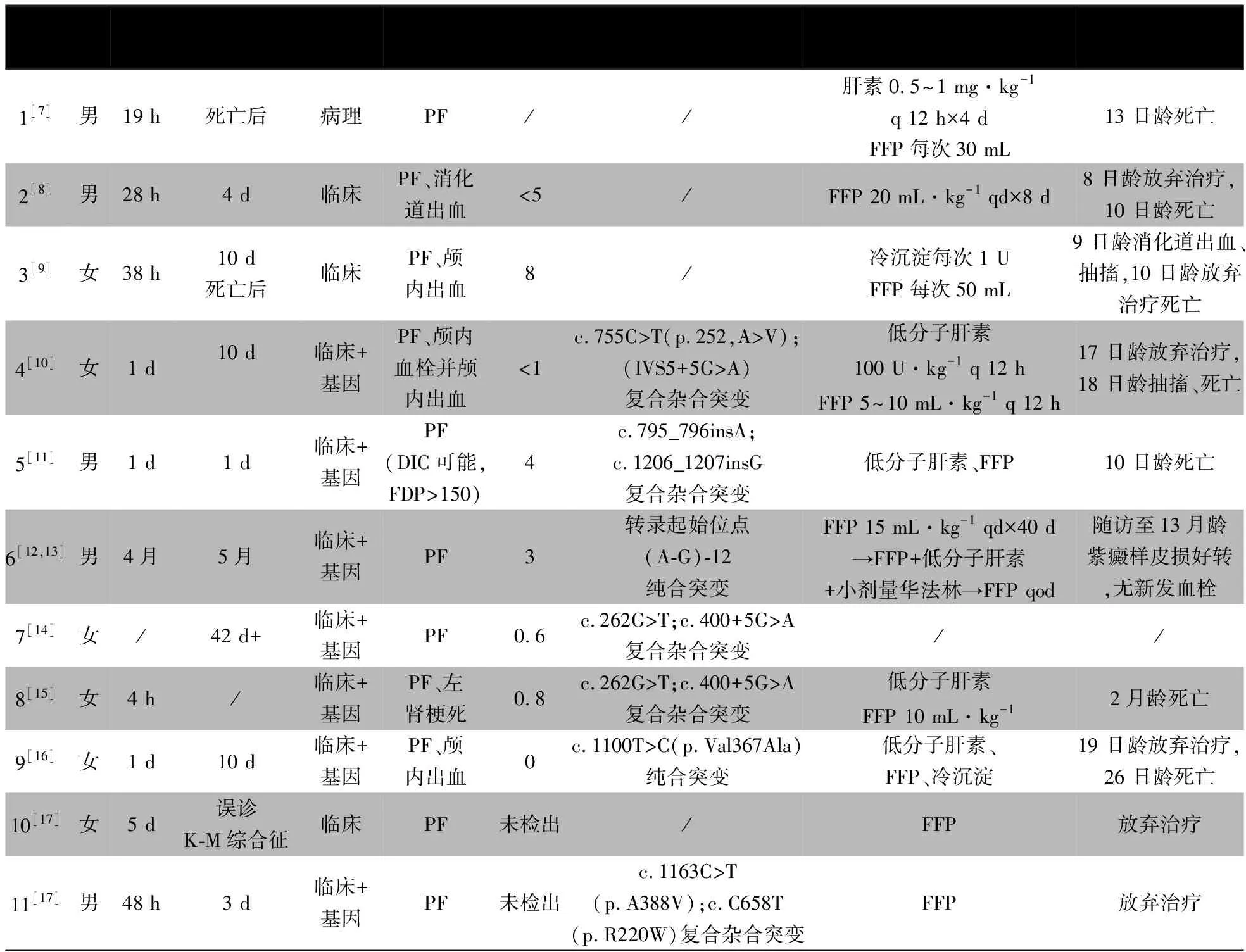
表2 文献报道中国新生儿期遗传性蛋白C缺乏症病例临床表现、诊断、治疗及预后
3 讨论
先天性PCD是一种罕见的遗传性易栓性疾病。PCD患儿蛋白C水平下降,无法通过抑制Ⅴa因子及Ⅷa因子发挥APC的抗凝作用[18],临床可表现为血栓事件的发生。复合杂合或纯合PROC突变的患儿表现为重度PCD(蛋白C<1%),国内报道的13例重度先天性PCD,12例新生儿期起病,10例于生后48 h内起病,说明重度先天性PCD起病急、进展快。新生儿的血浆蛋白C水平较低,6月龄时才达到正常成人水平。由于新生儿的生理性低维生素K,蛋白C不能进一步合成γ-羧基谷氨酸(Gla),从而进一步影响蛋白C发挥其抗凝作用,增加了与遗传性PCD相关的血栓事件的发病率及病死率[19]。本文文献复习中10例最终放弃治疗或死亡,说明新生儿期重度遗传性PCD病死率高,及时明确诊断并尽早开始治疗有助于挽救患儿的生命。
重度的遗传性PCD可在出生后数小时至几天内出现症状,本文文献复习中12例以PF为首发症状。表现为下肢、臀部、头部、躯干等全身多处出现紫癜样皮损,以下肢为著,皮损迅速扩大、融合,颜色由暗红逐渐进展为紫黑色并伴大疱形成,严重者甚至因为中等血管的栓塞进一步进展为局部坏死,演变为黑色焦痂,受累肢体可明显肿胀,甚至出现表面皮肤脱落。除皮肤表现之外,全身其他部位亦可因血栓栓塞而引起相关表现,有同时合并颅内血栓、出血,喂养不耐受,消化道出血,以及肾梗死的情况。更有甚者,重度PCD患儿在孕期即可出现多个器官的栓塞,甚至死于宫内[20]。
本文例2为国内首次报道以颅内和肺等重要脏器出血为首发表现,而无典型皮肤症状或血栓栓塞相关的表现。监测患儿凝血功能,在D-二聚体升高的同时,存在PT、APTT显著延长,Fbg下降,FDP明显升高,符合慢性DIC表现。可能是由于广泛的动静脉血栓栓塞,消耗凝血因子,产生继发性DIC,进而引起颅内出血等多脏器出血[21,22]。因此,遗传性PCD可以新生儿颅内出血、新生儿肺出血等出血性表现为首发症状,需要新生儿科医生提高此病的认识和警惕性。
PCD一经诊断,应立即干预,防止病情迅速进展而导致死亡。重度PCD患者于20世纪80年代初首次被报道,在蛋白C浓缩物被批准上市之前,多采用在急性期应用FFP或凝血酶原复合物(PCC)行替代治疗,至临床皮肤病变消除。维持期选择FFP进行长期替代治疗,或口服华法林长期抗凝治疗,防止血栓事件再发[23]。1991年首次有文献提出通过在急性期静脉输注蛋白C浓缩物在重度PCD的新生儿中进行替代治疗,使PF获得缓解[24],1996年首次有文献报道皮下注射蛋白C浓缩物使纯合突变的重度PCD患儿的蛋白C水平长期维持在安全值范围以上(>0.25 IU·mL-1)[25]。目前西方国家针对纯合或复合杂合突变的重度PCD的患儿,蛋白C浓缩物的替代治疗已成为主要的长期维持治疗方案。但在国内,由于蛋白C浓缩物价格昂贵,且并未上市投入临床使用,难以获得。本文文献复习中10例通过急性期的FFP替代治疗,辅以LMWH抗凝,取得了暂时的临床缓解。其中仅例6通过连续3月隔日输注血浆达到蛋白C替代治疗的目的,随访监测皮损改善、无新发血栓形成。值得注意的是,例6起病较晚,蛋白C水平偏高,对于其他重度PCD的患儿很难通过单纯血浆输注进行维持治疗。此外,由于重度PCD患儿多为新生儿,存在静脉穿刺困难等限制,而长期应用血浆进行替代治疗,需要面临反复静脉穿刺、血源制品供应越来越困难的问题,血源性感染如HBV、HCV、HIV等病毒感染风险也随着输血次数的增加而升高。不仅如此,由于蛋白C在FFP中的浓度较低,仅为4 μg·mL-1,往往需要较大剂量的FFP输注,以维持重度PCD患儿蛋白C水平在安全范围内,重复多次输注大量胶体,会引起高血压、高蛋白血症、肺水肿、蛋白尿等并发症。我院首次在国内尝试口服抗凝剂替代血浆作为长期维持治疗,以延长血浆输注间隔,减少其使用。
本文例1应用LMWH 5 d后开始重叠使用华法林,未再出现新发血栓症状。VKA可以通过降低Ⅱ、Ⅶ、Ⅸ和Ⅹ因子的活性抑制凝血,缓解PCD患者的血栓倾向,以重新达到体内出凝血的再平衡。目前已有重度PCD患者应用华法林作为长期治疗并取得疗效的报道[26-29],起始剂量0.15~0.4 mg·kg-1,直至调整至可以维持INR在2.5~3.5且D-二聚体正常的最低维持剂量,从而防止症状再发。
为保证华法林的安全性及有效性,需要面临长期监测INR的问题。对于严重遗传性PCD的新生儿,静脉穿刺难度高,反复穿刺亦有出血等风险。利伐沙班通过在内源性和外源性凝血途径中通过直接、选择性和可逆地抑制Xa因子(FXa)形成纤维蛋白凝块,从而抑制血小板活化,最终抑制凝血酶生成[30]。利伐沙班还具有可预测的药代动力学和药效学,出血事件发生率较低,无需频繁采血监测患儿的凝血功能。本文例2在家长充分知情同意的情况下,应用利伐沙班作为长期抗凝药,按需每1~2个月输注FFP,随访3年无再次发生严重瘀斑或出现新发生长迟缓,也没有受到任何药物不良反应的困扰。例2为国内首次,也是国际上最年幼的应用利伐沙班成功和安全地治疗PCD的病例。Menon等[31]报道了利伐沙班治疗13岁重度PCD的成功病例,Martinelli等[32]报道了出生时严重的蛋白S缺乏症(PSD)的6岁女童,在使用利伐沙班后没有出现新的皮肤坏死或血栓形成。表明利伐沙班可能作为一种有效的抗凝替代药物,用于严重先天性PCD或PSD的患者。
本文报告的2例PCD患儿经过3和6年的随访,均存活、生活质量尚好,且无明显不良反应。提示在急性期应用血浆替代治疗改善症状后,应用口服抗凝药物维持治疗,可明显减少或避免患儿长期血浆应用,且随访获得临床缓解、未影响生长发育。建议重度PCD的患儿,可考虑应用华法林或利伐沙班等口服抗凝剂作为长期维持治疗方案,从而改善其预后。
猜你喜欢
中国现代医生(2022年19期)2022-11-04
昆明医科大学学报(2022年4期)2022-05-23
中国典型病例大全(2022年7期)2022-04-22
中国听力语言康复科学杂志(2021年6期)2021-12-21
考试周刊(2017年26期)2017-12-12
校园英语·下旬(2017年7期)2017-07-14
科技视界(2016年27期)2017-03-14
中国实用医药(2016年29期)2016-12-26
中西医结合心血管病电子杂志(2016年17期)2016-11-17
上海医药(2016年11期)2016-06-30
