
氯倍他索丙酸酯的合成新工艺
2021-10-08 06:58王海波王瑞玲李合兴陈玉真
化学与生物工程 2021年9期
王海波,王瑞玲,李合兴,陈玉真
(河南利华制药有限公司,河南 安阳 455000)
氯倍他索丙酸酯为人工合成的高效局部外用糖皮质激素类药物,化学名为21-氯-9-氟-11-羟基-17-丙酸酯-16-甲基孕甾-1,4-二烯-3,20-二酮,具有较强的抗炎、抗瘙痒和毛细血管收缩作用,同时具有抑制细胞有丝分裂的作用,能有效地渗透进皮肤角质层;无水钠潴留作用,有一定的促进钠、钾排泄作用[1]。传统的合成工艺路线[2-3]是以9β,11β-环氧-17α,21-二羟基-16β-甲基-1,4-孕(甾)二烯-3,2-二酮(DB11)为起始物料,经9位上氟反应后,得到倍他米松;再在二甲基甲酰胺(DMF)体系中与原丙酸三乙酯进行大环反应,在乙醇-三氯化铝溶液体系中进行开环反应得到倍他米松-17-丙酸酯;然后在DMF体系中与甲基磺酰氯反应得到中间体,再与氯化锂反应,得到氯倍他索丙酸酯。该工艺路线较长,各步骤中间体分离操作繁琐,收率偏低,仅为71%左右;反应过程中用到剧毒品甲基磺酰氯,且在吡啶或DMF体系中进行氯代反应,产生大量高氨氮废水,不经济环保。因此,作者对该工艺路线各单元反应进行研究,调整产物结构构造的顺序,仍以DB11为起始物料,经过磺化-氯代-丙酰化、上氟、精制等反应合成氯倍他索丙酸酯(图1),其结构经1HNMR和ESI-MS确证。
1 实验
1.1 试剂与仪器
DB11,纯度98%以上,湖南新合新生物医药有限公司;其余所用试剂均为分析纯,安阳万光化玻有限公司。
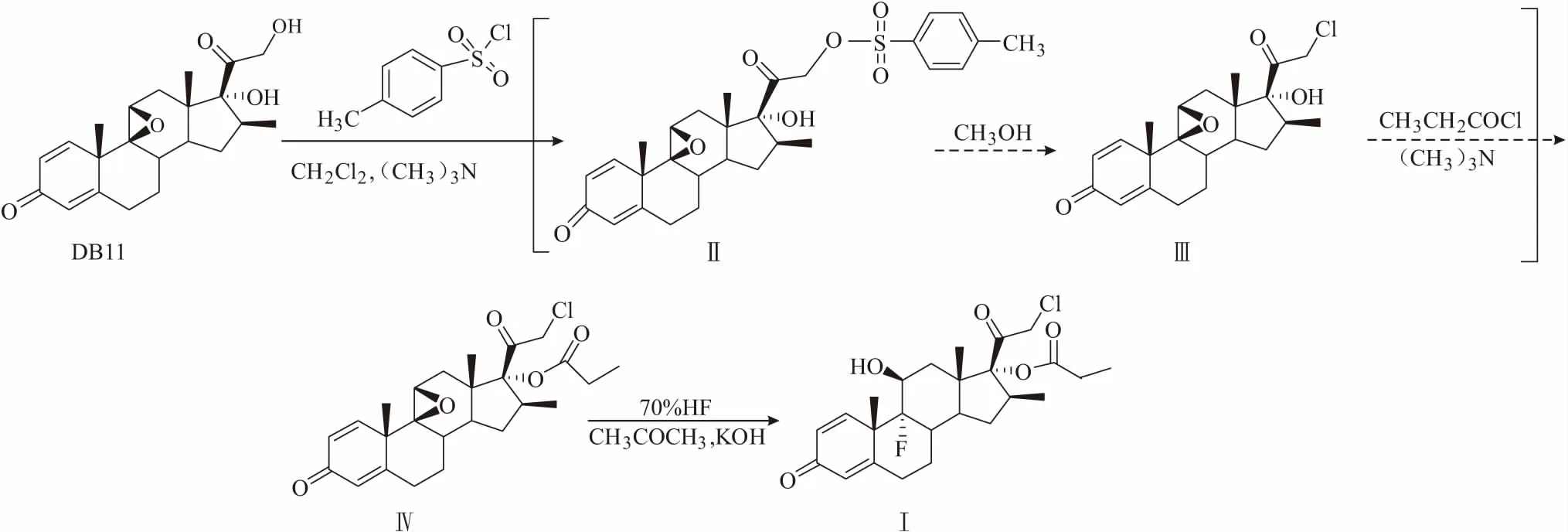
图1 改进的氯倍他索丙酸酯的合成工艺Fig.1 Improved synthetic process of Clobetasol propionate
YRT-3型熔点仪;Bruker AV-Ⅲ-500型核磁共振仪(CDCl3为溶剂);Agilent 1290-6540 UHPLC Q-TOF型液-质联用仪;Agilent 1260型高效液相色谱仪。
1.2 合成
1.2.1 中间体Ⅳ的合成[4-6]
在氮气保护下,将160 mL二氯甲烷、20 g DB11和20 g对甲苯磺酰氯加入反应瓶中,降温至-5~0 ℃,滴加20 mL三乙胺,30 min内滴完,于-5~5 ℃下反应5 h,TLC(二氯甲烷∶丙酮=6∶1)检测Rf0.35处几乎无斑点,得到中间体Ⅱ;向体系中加入2 mL甲醇,升温至30~35 ℃,保温反应10 h,TLC(二氯甲烷∶丙酮=6∶1)检测Rf0.65处几乎无斑点,得到中间体Ⅲ;在氮气保护下,将反应体系降温至-5~0 ℃,加入20 mL三乙胺,控制温度0~10 ℃,缓慢滴加丙酰氯15 mL,30 min内滴加完,0~10 ℃保温反应5 h, TLC(二氯甲烷∶丙酮=6∶1)检测Rf0.72 处几乎无斑点;向体系中加入100 mL饮用水,搅拌10 min,静置30 min分层,二氯甲烷层分入另一反应瓶中,水层用100 mL二氯甲烷洗涤,搅拌10 min,静置30 min分层,合并二氯甲烷层;减压浓缩,回收溶剂,加入40 mL甲醇夹带二氯甲烷,过滤,干燥10 h,得中间体Ⅳ23.10 g,收率96.37%,HPLC纯度98.65%,最大单杂0.28%。
1.2.2 氯倍他索丙酸酯(Ⅰ)的合成[7]
将200 g氢氧化钾加入到400 mL饮用水中,搅拌溶清,备用。将100 mL氢氟酸(70%HF)和10 mL丙酮加入到250 mL聚四氟乙烯反应瓶中,搅拌均匀,降温至-40~-30 ℃,控制温度在-40~-30 ℃内,1 h内加入20 g中间体Ⅳ,加完,控温-30~-20 ℃反应5 h。反应完毕后,将反应液缓慢加入400 mL饮用水中,滴加氢氧化钾溶液,调节pH值7.0~7.5,过滤,干燥,得氯倍他索丙酸酯粗品18.70 g,收率89.49%,HPLC纯度98.69%。
1.2.3 氯倍他索丙酸酯的精制
将18 g氯倍他索丙酸酯粗品加入90 mL甲醇和90 mL二氯甲烷中,搅拌升温溶清后加入1 g活性炭,回流30 min,过滤,滤液浓缩,抽滤,干燥10 h,得氯倍他索丙酸酯精品17.10 g,收率95.00%,HPLC纯度99.45%,最大单杂0.09%。
2 结果与讨论
2.1 中间体Ⅳ和氯倍他索丙酸酯的表征
中间体Ⅳ:m.p.201.3~204.5 ℃;1HNMR,δ:1.00(s,3H,H-18),1.14(t,3H,H-25),1.22(m,1H,H-15β),1.37(d,3H,H-20),1.54(m,1H,H-12α),1.55(s,3H,H-19),1.92(m,3H,H-7α),2.37(m,2H,H-24),2.50(m,1H,H-12α),2.64(m,1H,H-6β),3.94(d,J=15 Hz,1H,H-22b),4.04(d,J=15 Hz,1H,H-22α),4.44(m,1H,H-11),6.33(d,J=10 Hz,1H,H-2),7.21(d,J=10 Hz,1H,H-1);ESI-MS,m/z:446.45[M+H]+。
氯倍他索丙酸酯:m.p.197.5~199.3 ℃(194~198 ℃[8]);1HNMR,δ:1.00(s,3H,H-18),1.14(t,3H,H-25),1.22(m,1H,H-15β),1.38(d,3H,H-20),1.54(m,1H,H-12α),1.55(s,3H,H-19),1.60(m,1H,H-7β),1.93(m,3H,H-7α/15α/OH),2.01(m,1H,H-14),2.09(m,1H,H-16),2.37(m,2H,H-24),2.41(m,1H,H-6α),2.46(m,1H,H-8),2.49(m,1H,H-12α),2.64(m,1H,H-6β),3.94(d,J=15 Hz,1H,H-22b),4.04(d,J=15 Hz,1H,H-22α),4.43(m,1H,H-11),6.13(s,1H,H-4),6.34(d,J=10 Hz,1H,H-2),7.21(d,J=10 Hz,1H,H-1);ESI-MS,m/z:466.47[M+H]+。
2.2 讨论
在中间体Ⅱ的合成中,用对甲苯磺酰氯替代文献中用到的剧毒品甲基磺酰氯,反应条件更温和,反应更彻底,反应结束后加入少量醇类催化,可以与体系中的氯离子发生置换,生成中间体Ⅲ。传统工艺[9-10]中17位丙酰化反应是采用原丙酸三乙酯先进行大环反应,再开环水解,容易产生21位异构体杂质;本研究采用丙酰氯作为酰化试剂,反应更彻底,不会产生21位异构体杂质。收率及质量均有提高,实现了磺化-氯代-丙酰化三步反应在一个体系中完成,简化了工艺操作,缩短了合成周期。
在化合物Ⅰ的合成中,采用氢氧化钾溶液中和上氟体系,避免了传统工艺[11]中采用氨水中和而产生的大量高氨氮废水,以及采用碳酸钾溶液中和容易产生大量泡沫等问题,清洁环保。
3 结论
以DB11为起始物料,经过磺化-氯代-丙酰化、上氟、精制等反应合成氯倍他索丙酸酯,总收率81%以上,HPLC纯度99%以上。
该合成路线较传统合成路线具有以下优点:通过调整产物结构构造顺序,实现一锅法完成磺化-氯代-丙酰化三步反应,简化工艺操作,缩短了合成周期,避免了高氨氮废水的产生,减少了废水排放,符合绿色环保的要求。通过对工艺参数的优化,提高了产品质量,降低了生产成本,适用于工业化生产。
猜你喜欢
佳木斯大学学报(自然科学版)(2022年1期)2022-11-25
中国药学药品知识仓库(2022年10期)2022-05-29
中国典型病例大全(2022年7期)2022-04-22
化学工程师(2022年3期)2022-04-19
波谱学杂志(2021年3期)2021-09-07
粮油仓储科技通讯(2021年1期)2021-04-16
汕头大学学报(自然科学版)(2020年4期)2020-12-14
质量安全与检验检测(2019年6期)2019-02-11
科技与创新(2015年20期)2015-10-29
科技与企业(2015年20期)2015-10-21
