
一个单纯型甲基丙二酸血症家系的MUT基因突变分析
2021-02-23 04:45陈璐金一帮童郁潘丽琴李阳阳施建有戴显宁蔡晓晓许锴
浙江医学 2021年2期
陈璐 金一帮 童郁 潘丽琴 李阳阳 施建有 戴显宁 蔡晓晓 许锴
甲基丙二酸血症(methylmalonic aciduria,MMA)又称甲基丙二酸尿症,为常染色体隐性单基因遗传病,也是一种常见的有机酸血症,发病率一般为1/48 000~1/250 000[1]。该病主要是由于甲基丙二酰辅酶A变位酶(methylmalonyl-CoA mutase,MCM)自身缺陷或其辅酶腺苷钴胺素(Ado-Cbl)代谢缺陷所致[2],使甲基丙二酰辅酶A向琥珀酰辅酶A转换过程受阻;该酶的缺乏导致血液中甲基丙二酸及其相关有机酸增高,甲基丙二酸、甲基丙二酰肉碱、丙酸、甲基枸橼酸等中间代谢产物体内异常蓄积,进而引起肝、肾、脑等多脏器损伤,致死率、致残率高[3-4]。MMA临床表型及基因型复杂,至今已发现7种致病变异基因,分别是MMACHC、MUT、MMAA、MMAB、HCFC1、SUCLG1、SUCLA2 型,在我国以 MMACHC基因变异引起的MMA合并同型半胱氨酸血症最为多见,MUT基因变异引起的单纯型MMA次之[5]。笔者对1例单纯型MMA患儿进行早期干预诊疗,同时对该家系进行基因检测,以预防缺陷患儿再出生,现报道如下。
1 对象和方法
1.1 对象 MMA先证者为2018年12月温州市人民医院新生儿科收治的1例足月产男婴,气促5 h,呻吟不安,拟“呼吸困难,肌张力低下”收住入院。其父母此前生育过1例疑似MMA病男婴,出生10余天夭折。患儿入院后检验指标显示顽固性酸中毒PCO226 mmHg,HCO3-5.9 mmol/L;低血糖 0.7 mmol/L,高血氨 126 μmol/L,高乳酸16 mmol/L。颅脑MRI检查显示双侧枕部及天幕硬膜下血肿。对夫妇进行遗传咨询,双方非近亲婚配,否认有家族性遗传病史。本研究经本院医学伦理委员会批准,患儿家属知情同意。
1.2 方法
1.2.1 遗传代谢疾病检测 采集患儿指尖静脉血,滴于专用滤纸片上阴干送检。采用串联质谱仪(美国ABI公司API4000型)对共22种氨基酸、55种酰基肉碱进行检测。采集患儿尿滤纸片标本,安装新生儿尿袋留取新鲜尿液5ml,将尿液倒入无菌治疗碗中,将尿渗透滤纸浸润自然干燥后送检。采用气相色谱质谱仪(美国ABI公司API3200型)对尿液中的132种有机酸、氨基酸等代谢化合物进行检测。
1.2.2 DNA提取和全外显子测序 采集患儿静脉血2 ml于EDTA抗凝管中,用QIAampDNA Blood MiniKi(t德国Qiagen公司)试剂盒提取基因组DNA,Qubit 2.0(美国Thermo Fisher Scientific公司)进行DNA定量。取0.5 μg患儿样本DNA用超声仪(BioruptorNGS)随机打断成200 bp片段,用Truseq DNA Sample Preparation试剂盒处理两端连接测序接头构建文库,使用Roche Nimblegen SeqCap EZ v5.1试剂盒捕获全外显子区域,采用Illumina XTEN平台进行高通量测序分析查找可疑病因。
1.2.3 Sanger测序验证 针对致病MUT基因的突变位点所在外显子区域设计PCR上下游引物,以患儿及其父母的基因组DNA为模板进行PCR反应,用Sanger测序法进行验证。PCR反应产物电泳后由ABI3730完成一代测序;查看Sanger测序峰图(Chrmos软件),比对Sanger测序结果(SeqMan软件)展开验证。
1.2.4 突变致病性预测分析 对基因突变的致病性进行预测,将人类与其他种属的MCM氨基酸序列进行物种保守性比对分析。针对突变位点分析MUT基因突变前后由氨基酸变化引起的蛋白质空间结构的改变,初步阐明突变位点致病的可能机制。生物信息分析预测选用Polyphen-2、SIFT、Clustal X 和 Swiss-PdbViewer软件。
2 结果
2.1 遗传代谢疾病检测 患儿血丙酰基肉碱13.61 mol/L(参考值 0.35~4.20 mol/L),丙酰肉碱/乙酰肉碱 0.85 μM(参考值 0.03~0.20 μM);尿液检测甲基丙二酸为116.81 μM(参考值 0.2~3.6 μM),甲基枸缘酸为 3.65 μM(参考值0~1 μM),均显著高于正常参考值。
2.2 基因测序结果 本例患儿MUT基因为复合杂合突变:(1)c.1280 G>A(p.Gly427Asp)(编码区第 1280 号核苷酸由鸟嘌呤变异为腺嘌呤),导致氨基酸改变p.Gly427Asp(错义突变)。患儿母亲该位点为杂合变异,父亲无变异。(2)c.1677-1G>A(p.?)(外显子的最后一个碱基位于编码区1677位,该外显子相邻前方的内含子的第一个碱基由鸟嘌呤变异为腺嘌呤)为剪切突变,不属于多态性位点。患儿父亲该位点存在变异,母亲无变异。患儿及其父母MUT基因测序见图1。
2.3 基因突变的生物信息学分析 查询dbSNP、Hapmap、千人基因组计划等数据库,未发现MUT基因c.1280G>A具有多态性的可能。Polyphen-2软件对这个变异的预测分数为1,提示突变为有害突变(图2a),SIFT软件的预测分数均为-5.942(图2b),提示“有损害的”。Clustal X软件通过对5个人类同源物种的MCM蛋白进行保守性比对,结果 p.Gly427Asp高度保守(图3),提示这个位点具有重要的生物学功能。
2.4 突变结构和模型分析 应用Swiss-Pdb Viewer软件进行模型构建和突变后相互作用对比分析当非极性氨基酸Gly427突变为极性氨基酸Asp427后,Asp427侧链延长,增加一个与Thr78苏氨酸的氢键作用力(图4a、b)。
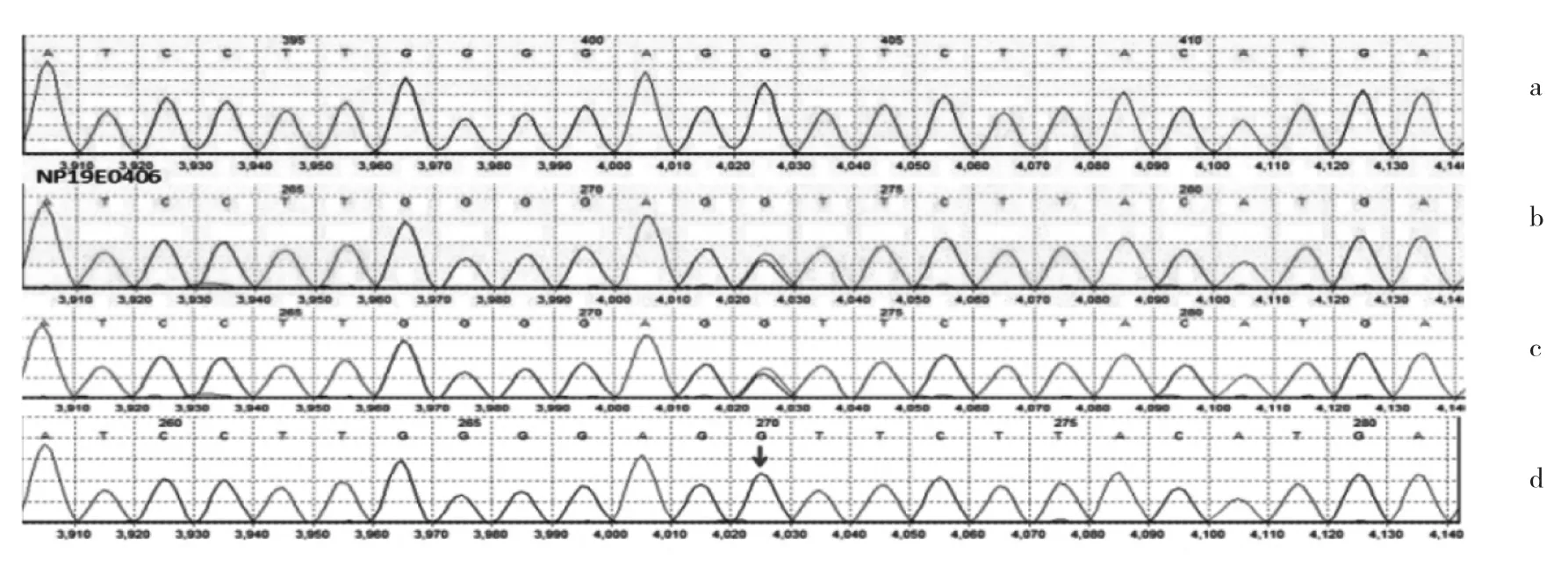
图1 患儿及其父母MUT基因部分序列测序结果(a、b、c、d分别是野生型序列、母亲、患儿和父亲测序结果)

图2 p.G427A突变致病性分析图(a:PolyPhen-2软件预测分数设定在0~1,预测分数越接近1,越可能为有害突变;b:PROVEAN软件预测结果有害的)
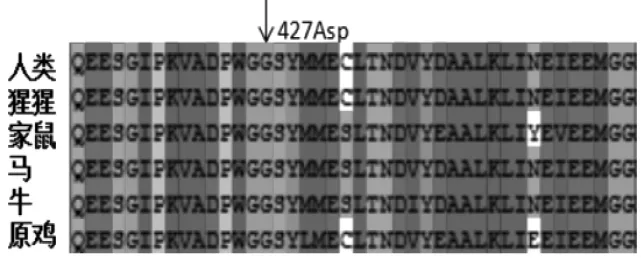
图3 人427位Gly与不同动物比较

图4 蛋白三维构象分析(a:427位氨基酸野生型;b:427位氨基酸突变型;虚线为氢键)。
3 讨论
MMA是一种常见的有机酸代谢遗传病,临床症状为代谢性酮症酸中毒,拒食、呼吸困难及生长发育迟缓等导致新生儿早期死亡或儿童期严重的神经功能障碍[6]。国内MMA患者60%~80%以MMACHA基因突变引起的合并型MMA居多,cblC缺陷是主要病因[7-8];其次为单纯型MMA,其中MUT基因是最常见突变类型[9]。MUT基因定位于6p21,全长约35 kb,内含13个外显子。MCM酶由MUT基因编码[10],是由两个相同亚基组成的同源二聚体,每个亚基包含32个氨基酸前导序列的750个氨基酸多肽链,进入线粒体后前导链立即被切除,装配成718个氨基酸的亚单位,每个亚单位包含两个主要的功能结合区:底物结合区(第86~423位氨基酸组成)和钴胺素结合区(第578~750位氨基酸组成),两个结合区之间被称为连接区(第424~577氨基酸组成),具有关闭8 barrel结构域的功能。第33~85氨基酸序列参与MCM亚单位二聚体化。本研究对1例经血液、尿液气相色谱/质谱串联检查高度怀疑为甲基丙二酸血症的新生儿家系进行了二代测序分析发现患儿患单纯型MMA,MUT基因检测存在两处变异,分别为第6号外显子G427A错义突变,第9号内含子c.1677-1G>A的剪切突变,为复合杂合变异。首先研究查询国内外相关数据库,排除这两个为多态性位点,用SIFT和Polyphen2软件进行G427A错义突变致病性分析为致病突变位点,并提供了氨基酸同源序列保守性分析,蛋白结构分析方法预测了突变位点带来的致病性改变。G427A的错义突变位于MCM酶结构连接区其改变了氨基酸的空间构象进而影响蛋白功能,这与Liu等[9]报道相符合,且该位点为我国南方常见突变位点[10-11]。其次,内含子剪切位点的突变可造成剪切位点破坏和识别异常,引起前体mRNA的剪切,从而造成基因的异常转录。根据剪切位点位置的不同对外显子所造成的剪切效应也不同,突变位于外显子上游-1bp和-2bp或+1bp和+2bp被称为是固有剪切位点,可破坏固有剪切位点,造成外显子完全不识别,最终导致蛋白异常表达影响蛋白功能,其致病性最强。第9号内含子c.1677-1G>A是典型的固有剪切突变,影响了mRNA的剪切[12],该剪切突变位点鲜有报道。本例通过气相色谱/质谱串联技术对先证者血液、尿液进行了检测高度怀疑为MMA,通过测序的方法准确作出MMA分型,位于MUT基因的两个突变位点 c.1280G>A(p.Gly427Asp)和 c.1677-1G>A(p.?)是导致单纯型MMA的主要原因,该类型属对维生素B12治疗无反应型,MMA基因型的分子生物学诊断有助于对不同类型MMA患者进行基因分型,准确选择诊疗方案;同时有助于进行家系携带者检测和产前诊断分析[13],为家庭优生优育提供具体可行的方向。分子生物学检测技术帮助临床医生提供精准治疗导向,切实对新生儿遗传代谢病做到早发现、早诊断、早干预、早治疗。
猜你喜欢
分子催化(2022年1期)2022-11-02
电子科技大学学报(2022年5期)2022-10-29
石油化工(2022年9期)2022-10-19
力学与实践(2022年3期)2022-07-02
南京医科大学学报(自然科学版)(2021年8期)2021-10-19
中国生殖健康(2020年4期)2021-01-18
航空发动机(2020年3期)2020-07-24
模具制造(2019年10期)2020-01-06
筑路机械与施工机械化(2015年1期)2015-09-18
湖北农业科学(2014年11期)2014-09-10
