
基于密度泛函理论的甲基芳烃液相氧化反应活性分析
2022-10-19 03:49周绪振吕全明孙伟振
石油化工 2022年9期
袁 方,周绪振,吕全明,孙伟振,赵 玲,3
(1. 华东理工大学 化学工程联合国家重点实验室,上海 200237;2. 江苏海伦石化有限公司,江苏 无锡 214400;3. 新疆大学 化学化工学院,新疆 乌鲁木齐 830046)
通过甲基芳烃液相氧化反应可将石油炼化的低值芳烃转变为高附加值含氧化合物,生成的芳香羧酸是聚酯和聚酰亚胺等高性能聚合物的单体[1-5]。甲基芳烃的液相氧化通常是在高温高压下进行的均相催化过程,遵循烃类液相氧化反应机理,可用链式反应机理来解释,包括链引发、链传递和链终止三部分[6-18]。甲基芳烃氧化的主要步骤(以甲基被氧化为醛基为例)包括:1)链引发阶段α-H 的夺取反应;2)链传递阶段甲基芳烃自由基的过氧化、α-H 夺取和过氧酸分解。随着苯环上甲基数量和位置的不同,甲基芳烃液相氧化所需要的温度和压力条件各不相同。然而,目前这一差异大多见于宏观的实验研究中,而基于微观层次(原子或分子尺度)的甲基反应活性研究很少有公开的报道。甲基芳烃的氧化大多在醋酸溶剂中进行。溶剂在有机化合物液相氧化过程中具有重要作用,主要影响自由基的链式反应进程[19-20]。因此,由于溶剂化的重要影响,在对甲基芳烃液相氧化过程的反应活性研究时,需要考虑合适的溶剂模型,以最大程度上符合实际液相氧化的特征。
本工作采用密度泛函理论(DFT)并考虑溶剂效应对反应的影响,针对甲苯(MB)、间二甲苯(MX)、对二甲苯(PX)、连四甲苯(PR)和均四甲苯(DR)液相氧化的反应机理进行过渡态搜索计算,通过比较不同甲基芳烃的液相氧化反应达到过渡态的活化能,分析不同甲基芳烃的反应活性。
1 计算方法
目前B3LYP,M06-2X,wB97XD 泛函是比较常用的准“万能泛函”,在计算速度和精度两方面均表现优异,是整体性能评价较好的计算泛函[21-25]。但B3LYP 不能描述范德华作用中的色散吸引部分,不但需要额外加上色散校正,且对里德堡激发和电荷转移激发的计算精度较差。2008 年诞生的M06-2X 和wB97XD 对B3LYP 的弱点有比较好的改进,但M06-2X 泛函对DFT 积分格点精度的要求远高于普通泛函,导致几何优化和自洽场(SCF)不容易收敛,或优化后出现小的虚频。wB97XD 和M06-2X 类似,都是给主族参数化,但计算含过渡金属的体系时会优于M06-2X,且适用于溶液反应。因此,本工作采用wB97XD 方法,在6-311G(d)基组上计算在反应溶剂中甲基芳烃液相氧化链引发阶段和链传递阶段所涉及的反应物、产物及过渡态的最优几何构型和分子能量,通过振动频率分析得到过渡态有且仅有一个虚频,通过内禀反应坐标的计算,确认过渡态两边正确的连接关系。本工作所涉及的量子化学计算,全部在曙光5000 型高性能计算平台上使用Gaussian09 程序包完成。
2 结果与讨论
2.1 链引发阶段α-H 的夺取
甲基芳烃液相氧化反应开始于不同价态催化剂组分之间的快速转化,其中Co 和Mn 充当氧化剂,Br 充当促进剂。Co(Ⅱ)首先在反应体系中被某些过氧化物氧化为Co(Ⅲ),后者迅速将Mn(Ⅱ)氧化为Mn(Ⅲ),自身转变为Co(Ⅱ);然后Mn(Ⅲ)氧化Br-生成Br·,本身变成Mn(Ⅱ),生成的Br·夺取甲基芳烃的α-H 来促进链引发[6,7,11,15]。模拟结果表明,在α-H 夺取过程中随着苯环上甲基数量的增多,过渡态构型中C—H 键长总体呈现增加的趋势,H—Br 键长总体呈现减小的趋势,说明甲基数量的增多有助于Br·对α-H 的夺取。这是因为甲基是供电子基,甲基数量的增多有助于苯环的共振稳定,形成稳定的CH2·,有助于Br·对α-H的吸引。比较具有相同甲基数量的芳烃分子可知,相比较于对位结构,间位结构对生成CH2·的位阻作用更大,表明后者苯环上的甲基更难被进攻,不利于Br·对α-H 的夺取。
几种甲基芳烃链引发阶段的反应能量变化如图1 所示。由图1 可知,甲基芳烃链引发阶段的活化能从高到低顺序依次为:MB>MX>PX>PR>DR,说明MB 上的甲基反应活性最低,低于二甲基芳烃的甲基反应活性,四甲基芳烃的甲基反应活性最高,且活化能显著低于二甲基芳烃和MB。这是因为甲基具有的供电子效应使多甲基苯环的共振稳定性更高[9],因而使得CH2·越稳定,α-H 越容易被夺取。与具有相同甲基数量的芳烃相比,二甲基芳烃的甲基反应活性差别较大,其中PX 的甲基反应活性大于MX 的甲基反应活性,说明间位上甲基的位阻效应较大。四甲基芳烃的甲基反应活性差别不大,其中DR 的甲基反应活性比PR 的甲基反应活性稍大,说明随着甲基数量的增多,不同取代基位置的甲基对α-H 活性的影响减小。另外,甲基的位阻和取代基的共同作用使得甲基数量增多时不同芳烃上甲基反应活性的差异逐渐减小。并且由能量变化可知,链引发阶段的α-H 的夺取反应是一个吸热反应。
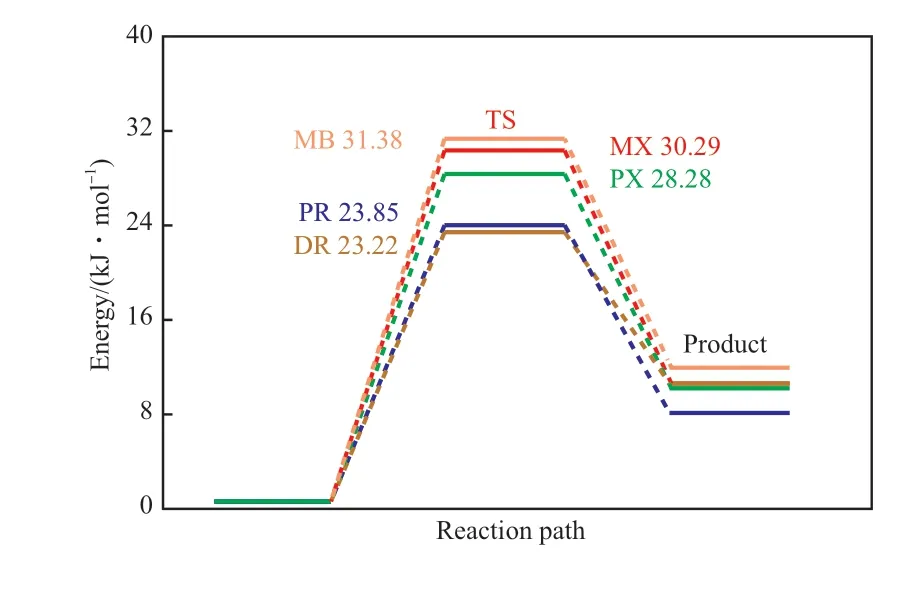
图1 甲基芳烃链引发阶段的过渡态活化能Fig.1 Transition state(TS) activation energy of chain initiation stage of methyl aromatics.
2.2 链传递阶段α-H 的夺取
在链传递阶段,自由基与氧的反应速率和过氧酸的分解速率往往很快,不是反应的决速步骤,过氧酸分解的反应速率常数通常比芳烃自由基(R·)与氧相互作用的速率常数小几个数量级。由于过氧自由基(ROO·)的浓度远大于R·的浓度,链传递的速率仅由过氧自由基和原始碳氢化合物之间的反应速率决定[19]。因此,链传递阶段的决速步骤是烷基过氧自由基对甲基芳烃上α-H 的夺取。
几种甲基芳烃链传递步骤的反应能量变化如图2 所示。由图2 可知,甲基芳烃链传递步骤的活化能从高到低顺序依次为:MB>MX>PX>PR>DR,说明MB 上的甲基反应活性最低,二甲基芳烃的甲基反应活性高于MB 的甲基反应活性,四甲基芳烃的甲基反应活性最高。但与链引发阶段有显著差异的是,不同甲基芳烃链传递步骤的活化能差值较小,且链传递的活化能远大于链引发阶段的活化能,说明甲基芳烃过氧自由基夺取α-H 的能力远小于Br·,这也是为什么采用Co-Mn-Br 三元催化剂的甲基芳烃液相氧化反应通常具有较高链引发效率的原因。由能量变化可知,链传递阶段α-H的夺取反应是一个吸热反应。
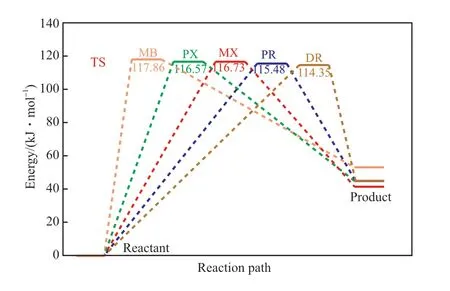
图2 甲基芳烃链传递阶段的过渡态活化能Fig.2 TS activation energy of chain propagation stage of methyl aromatics.
2.3 甲基反应活性估算
为了进一步明确甲基芳烃甲基反应活性的大小,本工作引入基于线性自由能关系的Hammett方程来定量表述苯环上的甲基反应活性大小[26]。所谓线性自由能关系是指A 系列反应的活化吉布斯函数(活化自由能)与B 系列反应中具有相同取代基的反应的标准摩尔吉布斯函数具有线性关系。Hammett 方程表达式见式(1)。

式中,k'为给定化合物转化的速率常数;k0为同样条件下MB 转化的速率常数;σ为苯环上取代基的特性常数;ρ为反应常数,取决于反应介质和温度。
以反应物苯甲酸和含取代基的苯甲酸为研究对象,通过分别测定苯甲酸及取代苯甲酸的离解常数,计算得到相应的Hammett 方程σ;Hammett取代基特性常数还列举了多种取代基的间位和对位的σ值。σ反映了苯环上间位和对位取代基对苯环侧链反应中心的影响程度,数值越小,苯环的侧链越容易参与反应(如甲基);相反,数值越大,苯环的侧链越稳定(如羧基)。
苯环上的取代基效应主要分为电子效应、场效应和空间效应3 类[27],电子效应又分为诱导效应和共振效应,包含贯穿共轭作用的σ大多包含了取代基效应中的电子效应和场效应,但并不包含空间效应。而苯环上的间位和对位的取代基离反应位点较远,空间效应对反应位点的影响极弱(除了一些分子量特别大的取代基),所以只有间位和对位的σ符合Hammett 方程的线性自由能关系。而对于苯环上的邻位取代基,即使分子量较小(H 除外)的甲基,空间效应对反应位点的影响都是不可忽略的。
尽管邻位上的取代基的空间效应不可忽略,但是目前尚未形成有效量化这一差异的方法。因此,本工作在计算甲基芳烃甲基反应活性的时候,借鉴Partenheimer 的处理方法[1],认为邻位取代基的影响和对位相同。甲基芳烃液相氧化反应大都在含水量为8%~10%(w)的HOAc 溶液中进行,在类似的实验条件下反应常数推荐为-0.95。σ数据来自于文献[28]。以MB 的氧化活性为基准(假定MB 的氧化活性为1),计算得到甲基芳烃的甲基反应活性关系如图3 所示。由图3 可知,当甲基芳烃的甲基被氧化为羧基后,剩余甲基的氧化活性显著降低;甲基数量越多,活性降低的越明显。对于二甲基芳烃来说,PX 初始的氧化活性为3.94,但是当第一个甲基被氧化为羧基之后,剩余甲基的氧化活性降低为原来的1/10。MX 初始的氧化活性低于PX,这是由于甲基是供电子基,且对位比间位对α-H 的作用更加显著,使得PX 的氧化活性大于MX。但当甲基被氧化为羧基后,由于羧基是强吸电子基,这时候对位的优势变为劣势,使剩余的甲基发生“钝化”,被氧化的难度增大。所以MX剩余甲基的氧化活性大于PX。四甲基芳烃的初始甲基氧化活性远远大于二甲基芳烃上的甲基,约为后者的5 倍左右。所以从理论上讲,相同条件下四甲基芳烃的氧化速率显著大于二甲基芳烃。随着甲基被依次氧化为羧基,剩余甲基的氧化活性开始显著降低,一般降低为原先的1/8 ~1/5 左右。可以发现,当四甲基芳烃只剩最后一个甲基的时候,甲基的氧化活性极低,仅为0.079。所以四甲基芳烃最终生成四甲基芳香酸的难度较大,所需的反应温度等条件更高,这在氧化实验中也得到了证实。另外,PR 和DR 的氧化活性除苯环上有两个甲基外基本一致,这说明随着甲基数量的增多,不同同分异构体之间的氧化活性差异趋于一致。

图3 甲基芳烃液相氧化甲基的氧化活性Fig.3 Methyl-oxidation activity of methyl aromatics in liquid-phase oxidation.
3 结论
1)芳烃上甲基的氧化活性从高到低顺序依次为:DR>PR>PX>MX>MB,且链引发和链传递阶段都具有相同的活性顺序。
2)在链引发阶段二甲基芳烃的甲基反应活性差别较大,而四甲基芳烃的甲基反应活性差别较小,表明随着甲基数量的增多,不同取代基位置的甲基对α-H 活性的影响减小,甲基的位阻和取代基的共同作用使得甲基数量增多时不同芳烃上甲基反应活性的差异逐渐减小。
3)在链传递阶段不同甲基芳烃过渡态的活化能差异较小,且链传递的活化能远大于链引发阶段的活化能,表明甲基芳烃过氧自由基的夺取α-H的能力远小于Br·。
4)通过Hammett 方程估算了甲基芳烃上甲基的相对反应活性,由于羧基是强吸电子基,当甲基芳烃的甲基被氧化为羧基后,剩余甲基的氧化活性显著降低,被氧化难度增大。
猜你喜欢
分子催化(2022年1期)2022-11-02
绿色科技(2022年14期)2022-08-12
科学家(2021年24期)2021-04-25
当代化工(2020年2期)2020-03-18
智富时代(2019年7期)2019-08-16
智富时代(2019年7期)2019-08-16
佛山陶瓷(2018年6期)2018-09-14
百科知识(2016年18期)2016-10-28
能源(2015年3期)2015-03-31
中学理科·综合版(2008年11期)2008-01-14
