
PDE4/PDE7双重抑制剂的虚拟筛选研究
2019-12-26 02:04陈喜陈慧慧陆婷婷赵新筠湛昌国
中南民族大学学报(自然科学版) 2019年4期
陈喜, 陈慧慧, 陆婷婷, 赵新筠, 湛昌国
(1中南民族大学 化学与材料科学学院,武汉430074;2 肯塔基大学 药学院,列克星敦,肯塔基州 40536)
磷酸二酯酶(PDEs)通过将第二信使分子cAMP和cGMP水解成无活性5′单磷酸核苷酸来调节机体的生理功能.PDEs由PDE1-PDE11等11个基因家族成员组成.其中,磷酸二酯酶4和7(PDE4和PDE7)主要分布于气管平滑细胞、炎症细胞及免疫细胞中.实验证明:PDE4酶抑制剂能够在有效范围内抑制大多数类型细胞参与炎症过程的各类体内功能性反应[1],用于治疗与炎症相关的疾病,如哮喘、关节炎和银屑病等[2-4].因此,PDE4酶是开发治疗慢性炎症药物的一个重要靶点.除了用于炎症有关的疾病上,PDE4抑制剂还被尝试用于其他一些医疗领域,例如刺激心肌收缩、抑制介质流失、防止血小板聚集、癌症化疗、治疗痛觉丧失、抑郁症、阿尔茨海默症、帕金森症及学习和记忆障碍等[5, 6].
尽管PDE4具有良好的抗炎作用,但PDE4抑制剂药物由于普遍存在恶心、呕吐等副作用,很大程度上限制了PDE4抑制剂作为治疗哮喘、COPD等慢性疾病的药物的使用剂量,削弱了其临床使用和治疗效果.有研究表明,这些副作用与PDE4D亚型被抑制有关[7].为了改善PDE4抑制剂作为抗炎药物的治疗效果,研究者提供了多种策略,其中采用多重PDE同工酶抑制剂的策略以其良好的可实践性备受瞩目.PDE7酶作为治疗慢性炎症的新靶点[8],受到越来越多的关注.研究证实,在PDE7被抑制的前提下,PDE4抑制剂的抑制炎症的作用得到了加强[8].因此,PDE4/PDE7双重抑制剂有望成为新一代的抗炎药物,用于治疗相关的疾病.
目前,PDE4/PDE7双重抑制剂报导比较少[9].随着高性能计算技术的进步以及药物设计理论的发展,分子虚拟筛选方法已被广泛应用于药物分子的发现研究[10].相比于传统的方法,它能够快速高效的从候选药物分子数据库中搜索结构新颖的潜在药物分子.为了寻找和发现新型的PDE4/PDE7双重抑制剂,本文联合运用包括OpenEye[11, 12]和AutoDock[13]在内的药物设计软件对自建的小分子数据库进行了大规模的虚拟筛选计算,结合相似性数据库搜索,初步获得了78个小分子化合物结构.并对这些分子进行了抑制活性测试,有24个分子具有显著的PDE4/PDE7抑制活性.最后对这些有活性的分子进行了定性的结构-活性关系分析,为PDE4/PDE7双重抑制剂的设计提供新颖的母体结构,可作为先导化合物用于后续的结构优化以及药物开发.
1 研究方法
本研究工作分为4个步骤,依次为提问结构的建立、分子虚拟筛选、分子对接以及抑制剂活性的测试.
1.1 提问结构的建立
选取6个对PDE4和PDE7均有较好的抑制活性以及选择性的小分子化合物[9],这些化合物的结构见图1,其对PDE4和PDE7的抑制活性数据列于表1中.首先,构建这些化合物的3D结构并进行能量最小化,以获得合理的分子构象.随后,使用AutoDock4程序[13]将6个抑制剂结构分别对接到PDE4B酶的活性位点(PDB: 3d3p)[14],对接过程中所用的参数均为程序默认.从对接产生的10个构象中选择结合自由能预测值最低的作为小分子的结构构象.最后,用OpenEye软件中的ROCs模块导入这6个抑制剂的结合构象,进行叠合及药效团分析,建立提问结构.X-射线单晶衍射结果表明,三种酶的结合口袋较为相似.以PDE4B的氨基酸排列顺序为例,三种酶结合口袋的亲水区均由位置高度保守的His238,His274,Asp275,His238,Asp392等氨基酸构成,而碱基结合口袋则由保守的氨基酸Phe414,Gln443,Phe448等氨基酸构成.据此,预期抑制剂在PDE4D,PDE7A中的结合模式与在PDE4B中的模式类似.因此,为简化研究步骤,未针对PDE4D,PDE7A酶再建立新的提问结构.
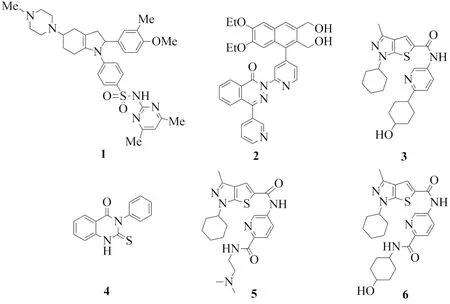
图1 PDE4/PDE7双重抑制剂的模板分子结构Fig.1 Structures of representative PDE4/PDE7 dual-specific inhibitors
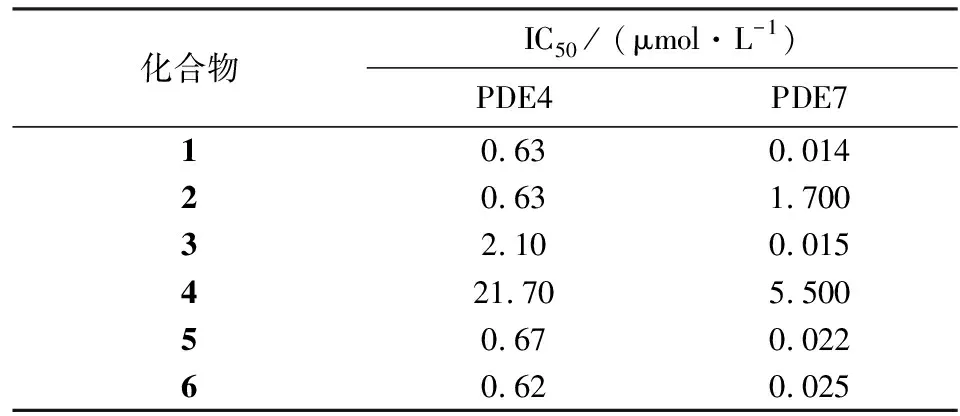
表1 代表性PDE4/PDE7双重抑制剂的抑制活性Tab.1 Inhibitory activities of representative PDE4/PDE7 dual inhibitors
1.2 分子虚拟筛选
此步骤工作主要利用OpenEye软件中的相应模块进行.首先利用Filter模块对自建的包含有5万个化合物结构的数据库进行预处理,筛除不符合成药条件的分子结构.使用OMEGA模块[12]对筛选出来的分子进行了构象搜索,建立相应的构象数据库.然后使用ROCs模块[11]在构象数据库中搜索与查询条件形状相似的分子结构,获得2036个小分子结构.最后,使用Fred模块将这些分子对接到PDE4B酶的结合位点(PDB: 3d3p)[14],根据打分筛选出70个化合物.
1.3 分子对接
使用AutoDock4程序将上一步筛选得到的70个对接到PDE4B蛋白质的活性位点(PDB: 3d3p),选取结合自由能预测值最低的构象进行分析,选择符合以下三个条件的分子结构.
(1)与PDE4B的结合自由能低于-7.0 kcal/mol;
(2)与Gln443氨基酸侧链存在氢键作用;
(3)具有芳香环且与Phe414和Phe446的苯环侧链存在π-π堆积作用.
通过以上分析,共获得了18个化合物结构.
1.4 活性测试
分析上述18个化合物的结构,提取出4个不同的公共结构.再从ChemicalBridge数据库中搜索包含这些公共结构的化合物,共得到78个小分子.随后,用酶联免疫法测试了这些化合物在10 μmol/L浓度下对PDE4B,PDE4D,PDE7A酶抑制活性.
2 结果与讨论
图2中列出了虚拟筛选的提问结构,该结构通过分析6个模板分子的对接构象获得.由图可知,提问结构具有5个药效团,分别为疏水基团(hydrophobic)、芳香环(ring)、极性基团(polar)、氢键受体(acceptor)以及氢键供体(donor).其中,芳香环、疏水基团以及极性基团为大多数模板分子所共有.使用ROCs模块以及提问结构对数据库进行形状相似性搜索以及对接筛选,最终命中18个化合物.根据公共分子骨架结构的不同,这些分子分属于4个分子系列1~4.不同分子系列的基本外形(shape1~4)以及药效团分布见图2.由图2可知:形状1和2具备了4个药效团,而形状3和4仅具备3个药效团.根据这4个系列化合物的公共结构,通过ChemicalBridge数据库[15]进行结构搜索,最终获得78个小分子化合物.生物活性测试结果表明,实验条件下这些化合物中有27个具有显著的酶抑制活性(抑制率大于50%),其结构-活性关系讨论如下.

图2 提问结构及命中的分子基本外形和药效团Fig.2 Shape andpharmcophore of the quiry structure and hit molecules
2.1 系列1化合物的结构和活性关系
表2列出了化合物系列1中各分子的结构及对PDE4B,PDE4D和PDE7A酶的抑制活性数据.比较各化合物对不同酶的抑制率数据,可见同一化合物对各个酶的抑制活性基本一致.以PDE4B酶的抑制率数据来讨论不同抑制剂的结构-活性关系.当R1的取代基为间位的羧基时,化合物的抑制活性均较高,进一步在旁边的邻位或对位引入不同的取代基并不会带来更高的抑制活性.此外,当R1取代基为对位的羧基时,分子的抑制活性也会较差. 当R2对位为氢、甲氧基或者Br时,化合物对三种酶的抑制活性均很好;当对位为氯原子时(化合物8,9和10),抑制剂则表现出一定的选择性;当间位引入各类取代基时,分子的抑制活性一般不会提高.由于分子中羧基的酸性较强,可预测在生理状态下该基团将处于带负电的状态.根据生物等排原理,对于此类化合物在R1取代基4-位引入带负电的基团,在R2取代基4-位引入体积较大的疏水性基团均有利于提高抑制剂的活性.
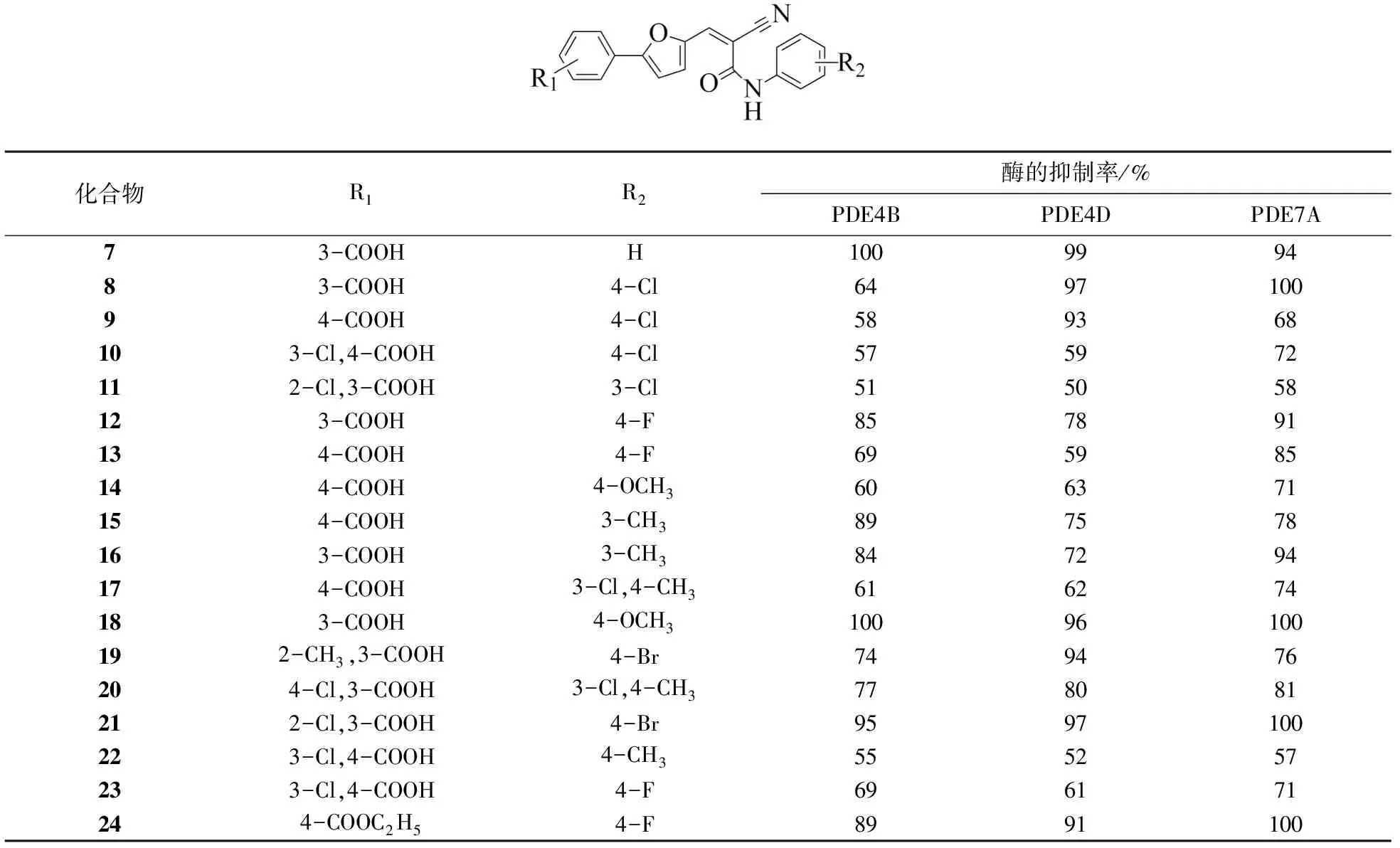
表2 系列1化合物对PDE4B,PDE4D,PDE7A酶的抑制率Tab.2 Inhibition rate against PDE4B, PDE4D and PDE7A for compounds from set 1
2.2 系列2化合物的结构和活性关系
该系列分子结构与模板分子1具有相似的苯基磺酰胺部分,且具有4个特征性药效团.尽管如此,系列中的分子酶抑制活性均不太高.原因可能是与苯基磺酰胺相连的酰基苯胺部分与三种酶的结合不好.由表3可见:当R1和R2为较大的卤素原子时(27),对PDE4B的抑制活性有一定的提高,而对PDE4D和PDE7A的抑制率较低.
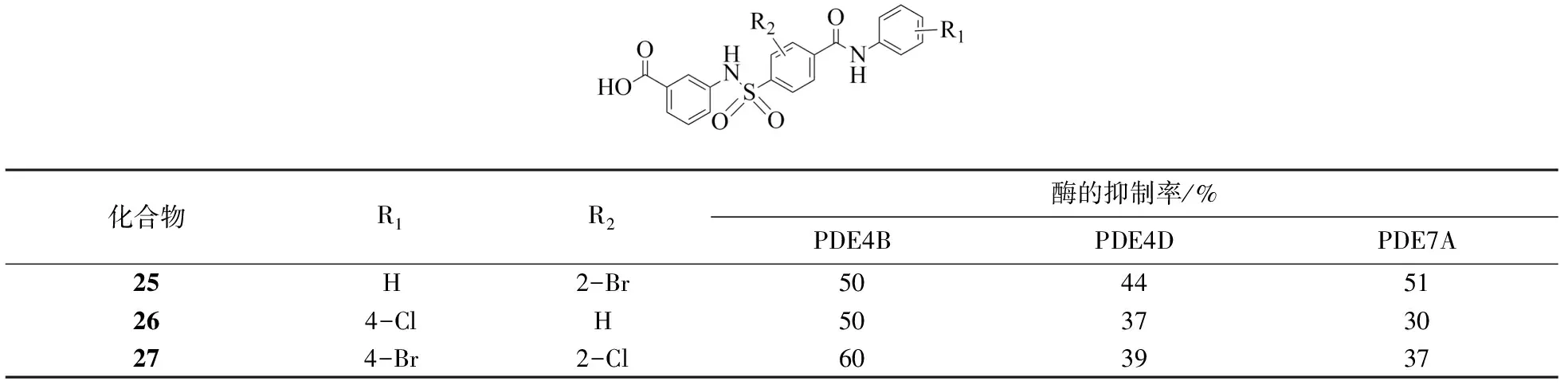
表3 系列2化合物对PDE4B,PDE4D,PDE7A酶的抑制率Tab.3 Inhibition rate againest PDE4B, PDE4D and PDE7A for compounds from set 2
2.3 系列3化合物的结构和活性关系
虽然该系列的分子结构只具备特征药效团中的3个,却表现出较好的生物活性.其中,化合物30对3种酶的抑制率均达到100%.通过分析其结构可知,当R取代基上疏水基团较小,且能提供一个氢键供体时,抑制剂的活性较好.对于化合物31,虽然其疏水基团-N-Pyrrolidine的大小与化合物29中对应的环戊基相仿,但缺乏氢键供体,因此活性略弱.此外,R上的疏水基大小对抑制剂的PDE7A抑制活性非常显著.例如,当R基团中的疏水基为环己基时(28),对PDE7A的抑制率仅为50%;当R基团的疏水基缩小为环戊基时(29),对PDE7A的抑制率则达到了100%.此系列化合物的公共结构与三类酶的天然底物cAMP的碱基和糖环部分结构类似,电荷分布也大致相同,也是造成该系列化合物活性高的成因.此外,公共结构的氮杂环上尚有空间可连接其他不同性质的取代基.因此,化合物30可以作为先导化合物的结构,通过化学修饰来获得活性及选择性更高的小分子.

表4 系列3化合物对PDE4B,PDE4D,PDE7A酶的抑制率Tab.4 Inhibition rate against PDE4B, PDE4D and PDE7A for compounds from set 3
2.4 系列4化合物的结构和活性关系
该系列化合物仅有两个成员(32和33),且活性均较弱.比较这两个化合物的取代基R1和R2,可见当两个基团中含有体积较大的疏水基团时,对三种酶的抑制率均较高.
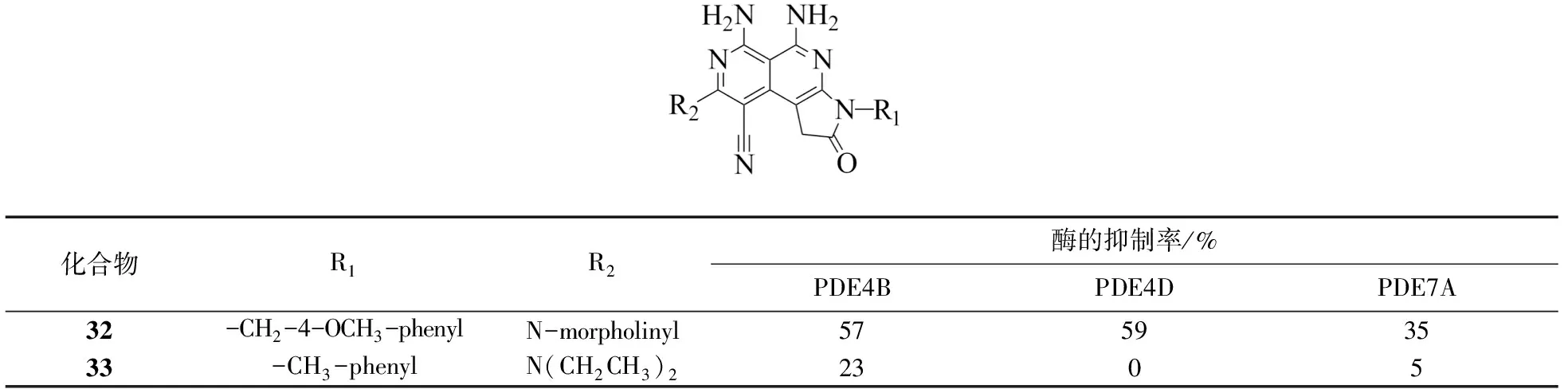
表5 系列4化合物对PDE4B,PDE4D,PDE7A酶的抑制率Tab.5 Inhibition rate againest PDE4B, PDE4D and PDE7A for compounds from set 4
总结上面4个系列化合物的结构-活性关系,可以发现命中的高活性化合物具备3个特征:具备不超过两个环的芳香中心;芳香中心左边有极性较大的取代基;芳香中心右边有体积较大且极性较小的非极性基团.
3 结语
本研究联合应用分子对接和相似性搜索的方法对自建的数据库进行了分子虚拟筛选研究,寻找新型的PDE4/PDE7双重抑制剂.通过分子搜索总共获得了78个小分子结构,分属于4个不同的化合物系列,这些化合物中有27个有显著的PDE4/PDE7酶抑制活性,有2类化合物结构可作为先导结构,用于高活性抑制剂分子的研发.
猜你喜欢
物理学报(2022年10期)2022-06-04
现代农村科技(2022年1期)2022-01-21
大连民族大学学报(2021年5期)2021-11-15
中国循证心血管医学杂志(2021年10期)2021-11-05
食品界(2021年7期)2021-07-19
商品与质量(2020年47期)2020-12-18
新课程·下旬(2019年7期)2019-09-17
发明与创新·中学生(2017年11期)2017-12-07
教育教学论坛(2017年14期)2017-04-20
创新科技(2014年10期)2014-07-27
