
原位阳离子交换法合成2D/0D SnS2/Ag2S异质结光催化剂及其还原Cr(Ⅵ)性能
2019-12-26 02:04胡军成邹思榕石义秋宋志周腾飞陈志伟
中南民族大学学报(自然科学版) 2019年4期
胡军成,邹思榕,石义秋,宋志,周腾飞,陈志伟
(1中南民族大学 化学与材料科学学院,武汉 430074;2武汉有机实业有限公司,武汉 430082)
有毒重金属离子六价铬Cr(Ⅵ)被广泛应用于电镀、颜料、抛光、制革、印刷等行业,由于大量的使用,使其成为了地下水和地表水的常见污染物之一.近年来,利用清洁、环保、经济的光催化技术将Cr(Ⅵ)催化还原为无二次污染的Cr(III),得到了广泛的研究[1]. 设计出新型、高效的半导体光催化材料是本领域的重点研究课题.在众多的半导体材料中,二硫化锡具有层状六方CdI2型晶体结构[2],具有带隙窄(2.2 eV)、可见光响应强、成本低、无毒、热稳定性好等优点,在降解甲基橙、罗丹明B等其他常见污染物中得到了关注和研究[3-6];但光生载流子易复合、量子效率低,导致催化效率较低.为了进一步改善二硫化锡的催化性能,提高光生电子和空穴的分离效率,常将SnS2与其他半导体材料复合构建异质结材料,如SnS2/TiO2、 GO/SnS2, BiOCl/SnS2和SnS2/SnO2等[4-9].
硫化银是一种窄带隙的直接半导体(Eg=0.9~1.1 eV),具有很宽的光响应范围[10],硫化银常作为共催化剂,与其它宽带隙半导体催化剂制备异质结复合材料,如ZnS/Ag2S,Ag2S/ ZnO和Ag2S/g-C3N4等[11]. 目前,关于SnS2/Ag2S异质结光催化的报道较少.由于SnS2的Ksp为2×10-27,Ag2S的Ksp为6.3×10-50,SnS2和Ag+之间可以进行原位阳离子交换,反应生成Ag2S. 因此,本文通过简单的两步水热法,原位阳离子交换合成2D/0D SnS2/Ag2S异质结,考察了其光催化还原Cr(Ⅵ)性能,并分析了催化剂的催化活性及稳定性.
1 实验部分
1.1 试剂和仪器
五水合四氯化锡SnCl4·5H2O、硫脲CH4N2S、硝酸银AgNO3、重铬酸钾K2Cr2O7、无水乙醇CH3COOH均为分析纯. X-射线粉末衍射仪(Bruker D8,德国 Bruker);场发射扫描电子显微镜(SU8010,日本 Hitachi);透射电子显微镜(Tecnai G 20,荷兰 FEI,200 KV);X-射线光电子能谱仪(VG Multilab 2000,美国 Thermal Electron);UV-Vis光谱仪(UV-2550,日本 Shimadzu);荧光光谱仪(F-7000, 日本 Hitachi).
1.2 催化剂的制备
SnS2的水热合成: 称取4 mmol SnCl4·5H2O和16 mmol硫脲分别溶于20 mL超纯水中,超声辅助溶解得到澄清溶液,在搅拌条件下充分混合后移至反应釜中,在180 ℃下进行水热反应20 h. 自然冷却至室温,用二次蒸馏水和无水乙醇交替洗涤样品去除杂质离子,60 ℃烘箱中干燥12 h.
原位阳离子交换合成2D/0D SnS2/Ag2S异质结:将前驱体SnS2六边形纳米片(0.6 mmol)分散于20 mL去离子水中,向所得悬浮液缓慢加入20 mL不同浓度的AgNO3(0.4, 0.8, 1.2 mmol·L-1)水溶液用于阳离子交换反应,样品分别被命名为H-0.4, H-0.8, H-1.2.将混合物溶液转移至100 mL聚四氟乙烯内衬中,在180 ℃下进行水热反应2 h,经洗涤干燥得到2D/0D SnS2/Ag2S异质结.
1.3 催化剂的性能测试
SnS2和SnS2/Ag2S异质结样品的光催化活性通过还原Cr(VI)来评价(350 W氙灯,λ>420 nm).在100 mL 光反应器中加入50 mg 催化剂及50 mL 20 mg·L-1的Cr(VI)溶液,避光黑暗条件搅拌1 h,确保建立吸附解吸平衡.在光催化还原过程中,间隔20 min从反应器中移取4 mL悬浮液,收集离心后获得的上清液. 根据二苯卡巴肼(DPC)法[12],用UV-Vis分光光度计测试其在540nm处的吸光度,定量分析Cr(VI)的光催化还原程度.
2 结果与讨论
2.1 晶体结构与形貌尺寸分析
图1为纯相SnS2纳米片及SnS2/Ag2S异质结样品的XRD谱图. 如图1a所示:
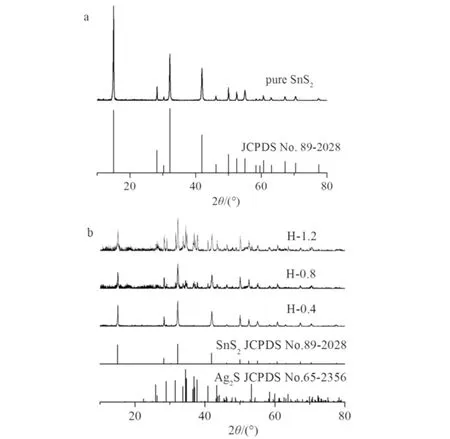
a)纯相SnS2纳米片;b) 所有SnS2/Ag2S异质结样品图1 纯相及SnS2/Ag2S异质结光催化剂的XRD谱图Fig.1 XRD spectrogram of pure SnS2 and SnS2/Ag2S heterojunction photocatalysts
所有衍射峰均被索引为六方晶系的SnS2(JCPDS No.89-2028),主衍射峰都很尖锐,且没有检测到其它的衍射峰,表明制备出了单相的SnS2且样品的结晶度较好. 从图1b中异质结(H-0.4, H-0.8, H-1.2)样品的XRD谱图可见:除纯相六方晶相的SnS2的衍射峰之外,其它衍射峰能很好地被归属为单斜晶系的Ag2S(JCPDS No.65-2356),随着AgNO3溶液浓度的增加,SnS2/Ag2S异质结样品中Ag2S的衍射峰的数目越来越多,且峰强越来越高,表明异质结中Ag2S的含量随AgNO3浓度的增加而增大.
纯相及异质结催化剂的SEM谱图见图2.由图2可知:一步水热反应获得的前驱体SnS2样品为均一的六边形纳米片,边长约为530 nm,且中心处有凸起. 在二次水热反应之后,所得的SnS2/Ag2S异质结样品(H-0.8)仍为均一的六边形纳米片,样品表面附着的一层均匀分布的小颗粒,边长约为600 nm,中心处的凸起仍然存在. 结合XRD推测,异质结样品表面的小颗粒为阳离子交换所得的Ag2S. 异质结样品基本保留了前驱体SnS2纳米片的形貌和尺寸,其三维结构由内部的2D结构SnS2和外层的0D结构Ag2S颗粒组成,且形貌均一.

a, b) 纯相SnS2六边形纳米片;c, d) 异质结样品H-0.8图2 纯相及异质结催化剂(H-0.8)的SEM谱图Fig.2 SEM images of pure SnS2 and heterojunction photocatalyst (H-0.8)
SnS2/Ag2S异质结样品H-0.8的TEM图见图3. 从图3a可见:所得异质结样品仍为六边形纳米片,与SEM结果相吻合. 图3c显示样品的晶面间距为0.31 nm,与SnS2的(100)晶面间距相一致.图3d表明样品的晶面间距为0.337 nm,与单斜晶系Ag2S的(012)晶面间距(0.338 nm)极为接近. 综合分析上述实验数据可知:Ag2S以小颗粒的形式附着在SnS2六边形纳米片的表面,形成均匀的2D/0D SnS2/Ag2S异质结光催化材料.

a)低分辨率图;b~d)高分辨率图(图c,d分别与图b中c,d框选中区域相对应)图3 SnS2/Ag2S异质结光催化剂(H-0.8)的TEM图Fig.3 TEM images of SnS2/Ag2S heterojunction photocatalyst (H-0.8)
2.2 化学组成和价态
纯相SnS2和SnS2/Ag2S异质结样品H-0.8的XPS谱见图4.对比XPS全谱图4(a)发现,异质结样品H-0.8的全谱图比纯相SnS2多了Ag 3d的峰. 图4b,4c,4d分别为Sn 3d, S 2p, Ag 3d的精细谱. 如图4b所示:纯样品中结合能为494.5 eV和486.0 eV处的峰分别为Sn 3d3/2和Sn 3d5/2,是典型的SnS2中Sn4+的特征峰;样品H-0.8中,结合能为495.0 eV和486.5 eV处的峰分别归属为Sn 3d3/2和Sn 3d5/2,是典型的SnS2中Sn4+的特征峰[13]. 相比纯相的SnS2, 样品H-0.8的Sn 3d峰均向高结合能的方向偏移了0.5 eV,这可能是由于Ag+的引入导致了Sn所处化学环境的改变.
图4c中纯相SnS2结合能为162.3 eV和161.1 eV处的两个峰分别对应S的2p1/2和2p3/2. 样品结合能为H-0.8中62.0 eV和160.9 eV处的两个峰归属为S 2p1/2和S 2p3/2[14]. 与纯相SnS2相比,S 2p的结合能向低方向移动,同样说明Ag+的引入引起了S所处的化学环境的改变. 图4d为Ag 3d的高分辨谱,高结合能373.4 eV对应的峰为Ag 3d3/2,低结合能367.4 eV处的峰为Ag 3d5/2[15]. 以上的XPS结果进一步证明,Ag+被成功引入异质结材料中.
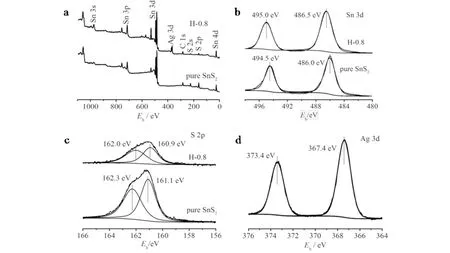
a) 全谱; b) Sn 3d; c) S 2p; d) Ag 3d图4 纯相SnS2和异质结光催化剂(H-0.8)的XPS谱图Fig.4 XPS images of pure SnS2 and heterojunction photocatalyst (H-0.8)
2.3 光学性能
为了测试样品的光吸收特征,对纯相SnS2和SnS2/Ag2S异质结样品H-0.8进行紫外可见固体漫反射测试(DRS).如图5所示:纯相SnS2样品在200~600 nm的波长范围内有明显的光响应;当形成SnS2/Ag2S异质结之后,样品H-0.8与纯相SnS2相比,其光响应范围明显拓宽,这一改变有利于提高光利用率,进而改善材料的光催化活性. 由公式(αhν) = A(hν-Eg)n/2计算可知,纯相SnS2的禁带宽度为2.27 eV,SnS2/Ag2S异质结样品H-0.8的禁带宽度为1.06 eV. 由文献[3]得知,Ag2S的禁带宽度约为0.96 eV. 对比发现,所得异质结样品H-0.8的禁带宽度介于纯相的SnS2与Ag2S之间.
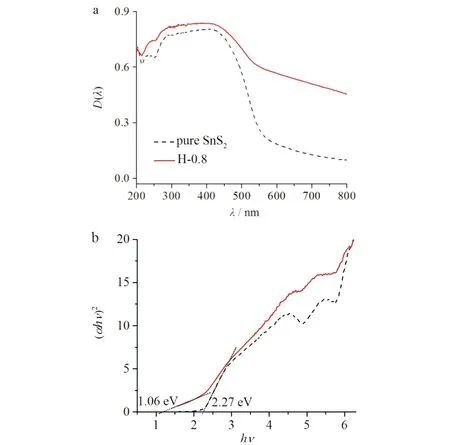
a)紫外可见固体漫反射;b)禁带宽度图5 纯相SnS2和异质结光催化剂(H-0.8)的光学性能图Fig.5 Optical performance images of pure SnS2 andheterojunction photocatalyst (H-0.8)
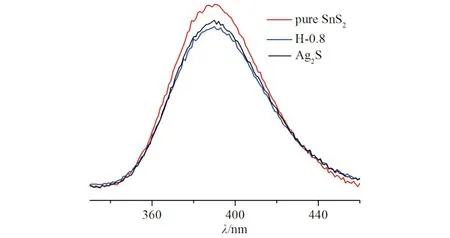
图6 不同光催化剂的荧光光谱(样品在240 nm处开始被激发)Fig.6 PL images of different photocatalysts
对所得样品进行室温下的稳态光致发光光谱表征(激发波长:365 nm),测试了材料在光照射时,光生电子空穴对的复合效率. 从图6可见:所有样品在350~450 nm波长区域展现发射峰,从谱图中可以发现,异质结样品H-0.8的发射峰强弱于纯相SnS2和纯相Ag2S,表明异质结催化剂H-0.8的光生电子空穴对的复合效率较低,有利于提高材料的光催化性能.
2.4 可见光下的催化活性
图7为不同样品对Cr(Ⅵ)的光催化还原性能图. 如图7a所示:Cr(Ⅵ)在可见光下的自还原几乎可忽略不计,表明Cr(Ⅵ)具有稳定性较好;与纯相的SnS2相比,SnS2/Ag2S异质结表现出明显增强的光催化活性.从图7b可见:可见光照射2 h后,样品的还原率分别为H-0.4 (51.7%),H-0.8 (59.4%),H-1.2 (58.1%), 纯相SnS2(42.9%),异质结样品H-0.8的光催化还原效果最好,并优于Cr(VI)还原的相关报道[16].随着AgNO3浓度从0 mmol·L-1增大到0.8 mmol·L-1时,SnS2/Ag2S样品(H-0.8)的光催化活性逐渐增强, 表明当反应生成Ag2S含量增大时,可扩大光响应范围,从而提高光生电荷的分离效率. 当AgNO3浓度增大至1.2 mmol·L-1,SnS2/Ag2S样品(H-1.2)活性略微下降,表明当Ag2S含量进一步提高时,过量的Ag2S会导致光生载流子的复合率升高[3].
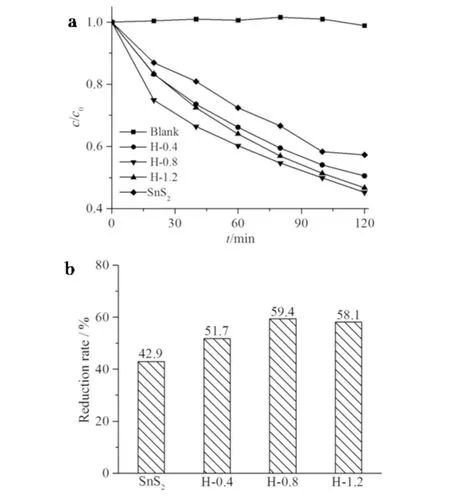
a)可见光(λ>420 nm)下不同催化剂对六价铬Cr(VI)的光催化还原曲线; b)相应的还原率直方图图7 不同光催化剂的光催化活性图Fig.7 Photocatalytic activity images of different photocatalysts
2.5 晶相稳定性和结构稳定性
在可见光照射下进行Cr(Ⅵ)催化还原实验之后,收集反应体系中的催化剂进行XRD及SEM表征,结果如图8和图9所示. 对比发现,实验后的XRD无杂质峰出现,出峰位置与实验前一致,表明反应后样品成份并未发生变化,晶相稳定性较好. SEM图显示样品为六边形纳米片,表面的小颗粒均匀分布,结构未损坏或坍塌,表明异质结样品H-0.8的结构稳定性良好.
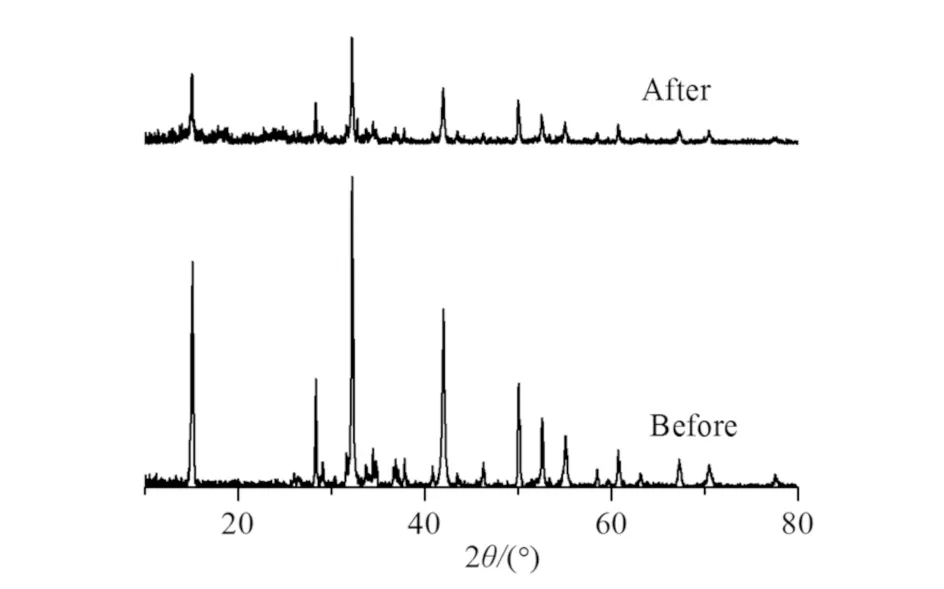
图8 光催化还原前后样品H-0.8的XRD图Fig.8 XRD Images of sample H-0.8 before and after photocatalytic reduction

a)还原前;b) 还原后图9 光催化还原前后样品H-0.8的SEM图Fig.9 SEM Images of sample H-0.8 before and after photocatalytic reduction
3 结语
通过简单的两步水热法成功合成了多级均一的2D/0D SnS2/Ag2S异质结材料. 二次水热条件下进行阳离子交换反应,生成的SnS2/Ag2S异质结基本保留了前驱体SnS2六边形纳米片的形貌和尺寸,其三维结构由内部的2D结构的SnS2纳米片和外层的0D结构的Ag2S小颗粒组成. Ag+的引入,明显提高了SnS2光催化还原Cr(Ⅵ)性能.异质结代表性样品H-0.8在可见光照射2 h下,光催化还原Cr(Ⅵ)效率达到59.4%,且材料具有良好的晶相稳定性和结构稳定性.合成的SnS2/Ag2S明显拓宽了SnS2的光响应范围,降低了光生电子空穴对的分离效率,提高了光催化活性.
猜你喜欢
科学之友(2022年11期)2022-11-03
湘潭大学自然科学学报(2022年2期)2022-07-28
辽宁石油化工大学学报(2021年6期)2022-01-04
无机盐工业(2020年1期)2020-12-31
科学导报(2020年70期)2020-11-09
消费导刊(2020年23期)2020-07-12
物理通报(2020年7期)2020-07-01
物理化学学报(2019年2期)2019-12-24
科学导报(2019年42期)2019-09-03
科技创新导报(2017年19期)2017-09-13
