
钯催化的串联环化反应构建β-内酰胺化合物
2019-12-26 02:04孙文武王硕张舒晗吴滨
中南民族大学学报(自然科学版) 2019年4期
孙文武,王硕,张舒晗,吴滨
(中南民族大学 药学院,武汉 430074)
β-内酰胺(2-Azetidinone)是一类含有四元氮杂环结构的化合物. 此类化合物具有良好的生物活性,广泛存在于药物分子和天然产物中. 除以青霉素为代表的抗生素外,β-内酰胺化合物在抗癌、抗病毒和抗高血糖等其他方面也展现出了优良的药理活性[1-5]. 同时,β-内酰胺作为一种重要的合成中间体,越来越多地应用于合成氨基酸、多肽及其他天然和非天然活性化合物[6-8]. 目前随着抗生素的大量使用,细菌的耐药性明显增加,对于β-内酰胺化合物种类多样性的需求也相应不断提高[9-11]. 所以,发展简便高效的方法来构建β-内酰胺一直是有机合成研究的热点.
合成β-内酰胺的方法包括分子内缩合、Staudinger反应和插羰基化反应等. 随着金属有机化学的发展,利用过渡金属催化碳氢键活化反应构建此类化合物的方法被相继报道[12]. 例如,以吡啶基异丙胺和8-氨基喹啉作为导向基团,实现了Pd(Ⅱ)催化的C(sp3)-H键活化的分子内胺化反应,立体选择性地合成了手性的α-氨基-β-内酰胺衍生物[13,14]. 本课题组利用酰胺为底物,醋酸钯作为催化剂,以五氟碘苯作为一个奇特的氧化剂,高区域选择性地合成了一系列单环以及双环的β-内酰胺化合物[15,16]. 但是,已报道的方法存在导向基团不易离去、底物局限性较大的问题. 同时,直接以化合物1a为底物合成3a类β-内酰胺化合物的方法还未见报道. 本文以N-(8-喹啉基)丙酰胺为原料,发展钯催化的碳氢键活化的串联环化反应,一步合成β位芳基取代的β-内酰胺(图1).

图1 一步合成β-内酰胺Fig.1 One-step synthesis of β-lactams
1 实验部分
1.1 仪器与试剂
微波反应器用CEM探索者SP系统;1H-NMR 和13C-NMR 在 Bruker Avance III 600 核磁共振仪上测定,所用氘代试剂均为 Cambrige生产,TMS 作为内标,δ 单位为ppm,J单位为 Hz. 层析用硅胶(300~400目)以及制备薄层板(厚度 0.4~0.5 mm)均为烟台江友公司生产. 金属催化剂、芳基碘苯、1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐(EDCI) 和4-二甲氨基吡啶(DMAP)等均购于Alfar Aesar公司.
1.2 原料制备
羧酸(1.0 equiv)和8-氨基喹啉(1.2 equiv)溶于二氯甲烷,随后加入EDCI(1.2 equiv)和DMAP(0.1 equiv). 混合物在室温下搅拌24 h,随后加入二氯甲烷,用1 mol/L的稀盐酸水溶液洗涤3次,再用盐水洗涤1次,有机相用无水硫酸钠干燥,浓缩后用柱层析纯化,干燥称重得到目标化合物.
1.3 β-内酰胺制备
在10 mL的微波反应管中依次加入酰胺底物(1 equiv),碘苯(1.2 equiv),Pd(OPiv)2(10 mol%),AgCO3(3.0 equiv) 和五氟碘苯(5.0 equiv). 首先把反应混合物在室温下搅拌大约5 min,然后放在微波反应器上. 控制程序升温:用20 W的功率加热到50 ℃反应1 min;再次用50 W的功率把反应温度从50 ℃升到120 ℃,保持这个温度反应3 min,最后用100 W的功率加热到150 ℃,在该温度下反应2 h. 待温度冷却到50 ℃时开盖拿出反应管,用薄层层析板分离得到目标化合物.
2 结果与讨论
2.1 反应条件的优化
首先选用酰胺化合物1a与对甲基碘苯2a作为底物,以Pd(OAc)2为催化剂,加入3倍当量的AgOAc和5倍当量的五氟碘苯在无溶剂体系下于微波反应器中加热到150 ℃反应2 h,分离得到39%的β-内酰胺化合物3a和8%的酯化产物4a(表1,entry 1),反应流程如图2. 随后以AgCO3代替AgOAc,看是否可以抑制化合物4a的生成,提高目标化合物3a的收率.3a的收率稍微提高,但是副反应没有被抑制(表1,entry 2). 进一步以PdCl2和Pd(OPiv)2为催化剂代替Pd(OAc)2参与反应,目标化合物的收率达到了64%,没有4a类酯化产物生成(表1,entry 3~4). 利用油浴代替微波加热,反应时间为24 h,β-内酰胺化合物3a的收率为55%(表1,entry 5). 至此确定了最优的反应条件:酰胺底物1a与对甲基碘苯2a在10 mol% Pd(OPiv)2,3.0倍当量的AgCO3,5.0倍当量的五氟碘苯存在下,通过微波反应器加热到150 ℃反应2.0 h可以达到最佳的反应效果.
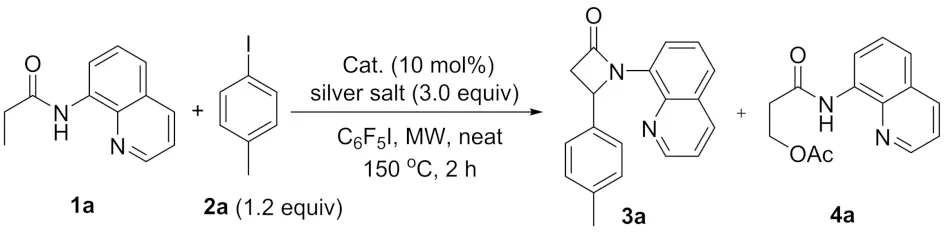
图2 催化剂和银盐的考察Fig.2 Survey of catalysts and silver salt

表1 反应条件的优化Tab.1 Optimization of the reaction conditions
aIsolated yield,bthe reaction was heated in an oil bath for 24 h
2.2 反应底物的扩展
在得到了最优反应条件之后,对底物的普适性进行考察. 如图3和表2所示,首先选用N-(8-喹啉基)丙酰胺为底物,与苯环上带不同取代基的碘苯反应. 在该反应条件下,苯环上具有不同取代基团的碘苯都能发生反应,官能团的兼容性较好. 以碘苯2b为偶联试剂,化合物3b的收率为62%,对比模板底物2a,产物的收率没有发生明显变化.但是当以具有吸电子取代基团的碘苯为底物时,目标化合物的收率显著降低,如卤素取代的化合物3c、3d和3e的收率分别为32%、31%和24%. 在标准条件下,以26%的分离收率得到氰基取代的化合物3f.
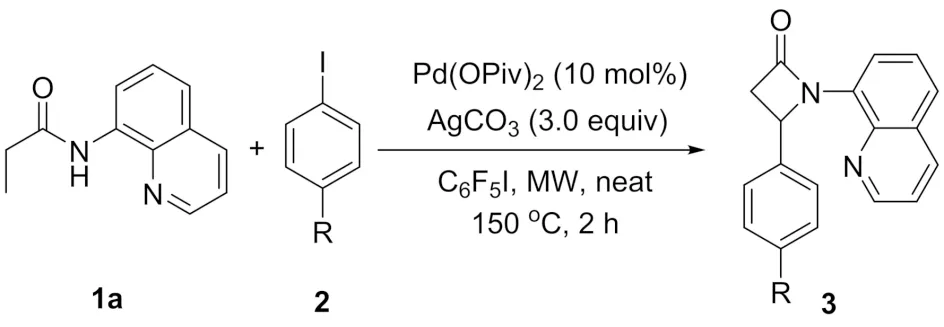
图3 碘苯的范围Fig.3 Scope of the iodobenzenes
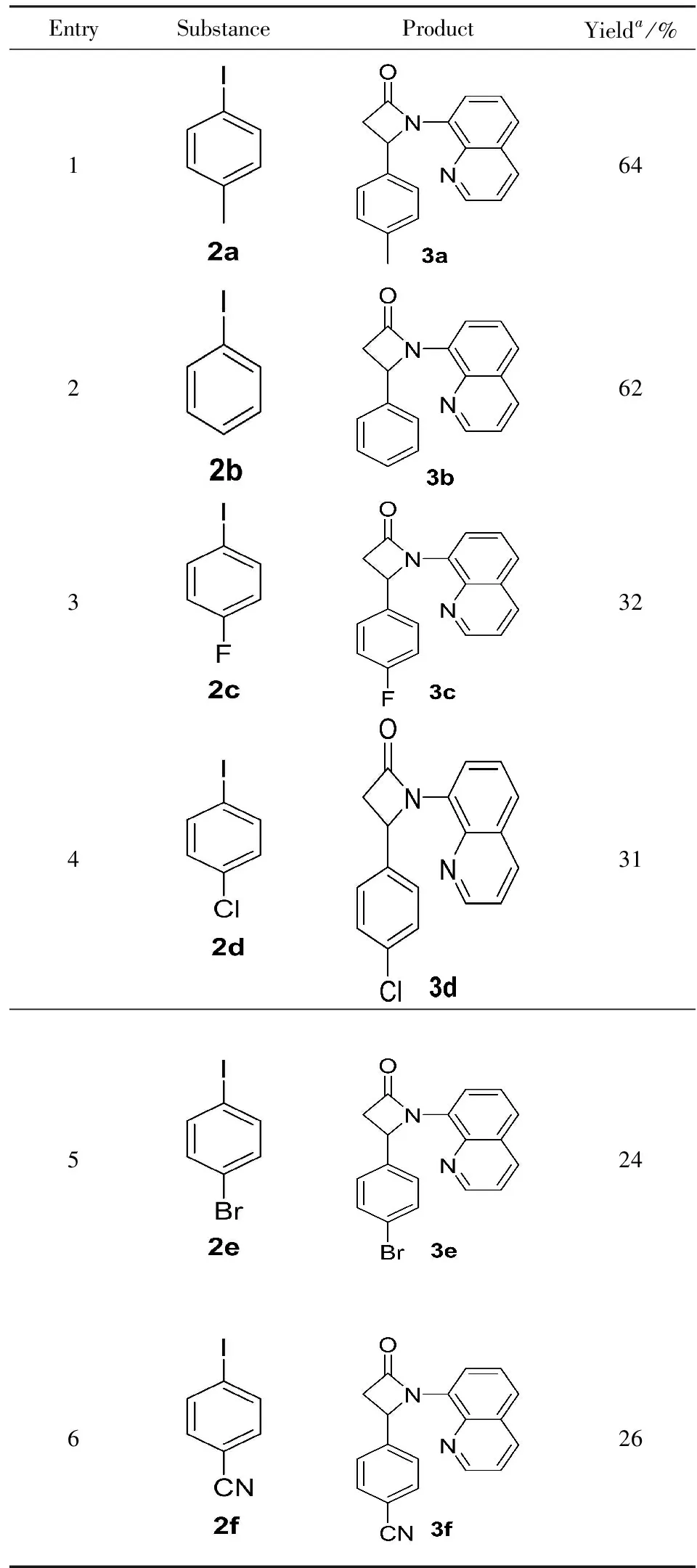
表2 底物的扩充Tab.2 Substrate scope
aTypical reaction conditions: substrate1a(0.10 mmol),ArI(1.2 equiv),Pd(OPiv)2(10 mol%),AgCO3(3.0 equiv) and C6F5I(5.0 equiv) in microwave at 150 ℃ for 2.0 h. Isolated yields
2.3 反应机理
通过对文献进行总结[13-17]并结合上述的实验结果,可能的反应机理如图4所示,首先N-(8-喹啉基)丙酰胺1a与Pd(II)配位,随后活化β位的碳氢键生成络合物A,接着与碘苯氧化加成生成四价的络合物B,然后还原消除得到芳基化产物C,此化合物进一步碳氢键活化生成中间体D,C6F5I与此中间体氧化加成形成四价的钯络合物E,再经过还原消除生成目标化合物3a.
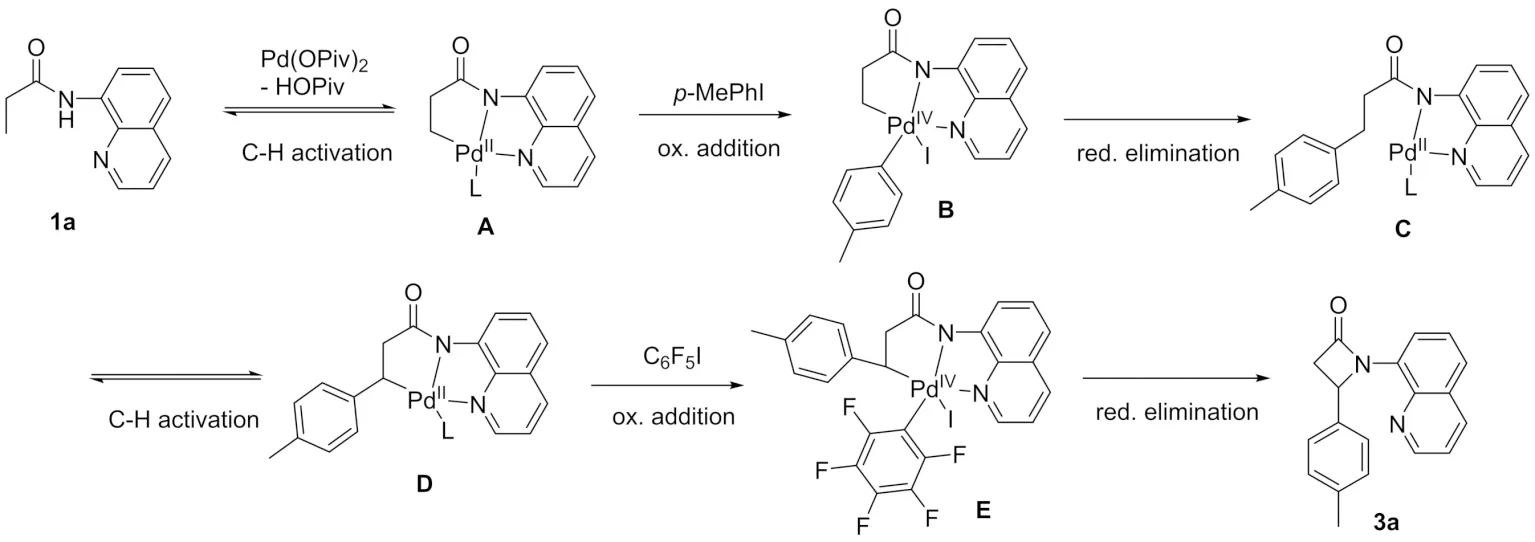
图4 可能的反应机理Fig.4 Plausible reaction mechanism
2.4 产物的谱图数据
1-(8-Quinolinyl)-4-(4-tolyl)-azetidinone(3a)[15]:1H NMR(600 MHz,CDCl3)δ8.77(dd,J=4.2 and 1.8 Hz,1H),8.27(dd,J=7.3 and 1.6 Hz,1H),8.00(dd,J=8.3 and 1.8 Hz,1H),7.58-7.46(m,2H),7.34-7.29(m,3H),7.02(d,J= 7.9 Hz,2H),6.23(dd,J=5.6 and 2.6 Hz,1H),3.69(dd,J=15.1 and 5.6 Hz,1H),3.08(dd,J=15.1 and 2.6 Hz,1H),2.22(s,3H).
1-(8-Quinolinyl)-4-phenyl-azetidinone(3b)[15]:1H NMR(600 MHz,CDCl3)δ8.76(d,J=4.2 Hz,1H),8.29(d,J=7.1 Hz,1H),8.02(d,J=8.3 Hz,1H),7.64-7.35(m,4H),7.35-7.07(m,4H),6.26(d,J=4.7 Hz,1H),3.72(dd,J=15.2 and 5.6 Hz,1H),3.10(d,J=15.2 Hz,1H).
1-(8-Quinolinyl)-4-(4-fluorophenyl)-azetidinone(3c)[15]:1H NMR(600 MHz,CDCl3)δ8.74(dd,J=4.2 and 1.8 Hz,1H),8.30(dd,J=7.4 and 1.5 Hz,1H),8.03(dd,J=8.3 and 1.8 Hz,1H),7.56-7.45(m,2H),7.38-7.35(m,2H),7.31(dd,J=8.3 and 4.1 Hz,1H),6.90(t,J=8.7 Hz,2H),6.25(dd,J=5.7 and 2.6 Hz,1H),3.71(dd,J=15.2 and 5.6 Hz,1H),3.07(dd,J=15.2 and 2.7 Hz,1H).
1-(8-Quinolinyl)-4-(4-chlorophenyl)-azetidinone(3d)[15]:1H NMR(600 MHz,CDCl3)δ8.72(dd,J=4.2 and 1.8 Hz,1H),8.32(dd,J=7.4 and 1.6 Hz,1H),8.03(dd,J=8.3 and 1.8 Hz,1H),7.64-7.43(m,2H),7.33(d,J=8.5 Hz,2H),7.30(dd,J=8.3 and 4.1 Hz,1H),7.18(d,J=8.5 Hz,2H),6.25(dd,J=5.7 and 2.7 Hz,1H),3.71(dd,J=15.1 and 5.7 Hz,1H),3.06(dd,J=15.1 and 2.7 Hz,1H).
1-(8-Quinolinyl)-4-(4-bromophenyl)-azetidinone(3e)[15]:1H NMR(600 MHz,CDCl3)δ8.72(dd,J=4.1 and 1.8 Hz,1H),8.33(dd,J=7.4 and 1.5 Hz,1H),8.03(dd,J=8.3 and 1.8 Hz,1H),7.57-7.48(m,2H),7.34(d,J=8.5 Hz,2H),7.31(dd,J=8.3 and 4.2 Hz,1H),7.29-7.25(m,2H),6.24(dd,J=5.6 and 2.6 Hz,1H),3.71(dd,J=15.1 and 5.7 Hz,1H),3.06(dd,J=15.2 and 2.7 Hz,1H).
1-(8-Quinolinyl)-4-(4-nitrilephenyl)-azetidinone(3f)[15]:1H NMR(600 MHz,CDCl3)δ8.85-8.70(m,1H),8.41(dd,J=7.3 and 1.6 Hz,1H),8.04(dd,J=8.3 and 1.8 Hz,1H),7.60(d,J=8.2 Hz,2H),7.56-7.47(m,2H),7.41(d,J=8.2 Hz,2H),7.30(dd,J=8.3 and 4.2 Hz,1H),6.33(dd,J=5.7 and 2.7 Hz,1H),3.75(dd,J=15.2 and 5.8 Hz,1H),3.06(dd,J=15.2 and 2.7 Hz,1H).
3 结语
发展了一种以Pd(OPiv)2为催化剂,8-氨基喹啉为导向基团,通过分子内碳氢键活化胺化反应一步合成β-内酰胺的新方法. 反应经历了两次碳氢键活化过程,第一次通过分子间的偶联生成芳基化的产物,第二次通过分子内的环化反应生成目标化合物. 反应具有较好的官能团兼容性,但是取代基效应对产物的收率影响较大,具有吸电子取代基团的碘苯收率明显降低.
猜你喜欢
当代医药论丛(2022年21期)2022-12-21
中国防痨杂志(2022年7期)2022-11-25
河北师范大学学报(自然科学版)(2022年5期)2022-09-20
中国现代中药(2022年4期)2022-05-08
纺织检测与标准(2021年1期)2021-12-05
波谱学杂志(2021年3期)2021-09-07
食品安全导刊(2021年20期)2021-08-30
——以高中化学“氢键”的教学为例
教学月刊(中学版)(2020年13期)2020-12-29
中国食品(2020年18期)2020-10-15
生物工程学报(2020年1期)2020-03-12
