
流动注射法测定水中挥发酚的不确定度评估
2019-05-28 02:12高倩倩向元英周竞达
云南化工 2019年3期
曾 锐,高倩倩,向元英,周竞达
(德阳市环境监测中心站,四川 德阳 618000)
酚是一种中等强度的化学毒物,与细胞原浆中的蛋白质发生化学反应。低浓度时使细胞变性,高浓度时使蛋白质凝固。酚类化合物可经皮肤粘膜、呼吸道及消化道进入体内。低浓度可引起蓄积性慢性中毒,高浓度可引起急性中毒以致昏迷死亡。根据酚类能否与水蒸气一起蒸出,分为挥发酚和不挥发酚。而挥发酚通常是指沸点在230℃以下的酚类,通常为一元酚。流动注射法测定水中挥发酚与传统方法相比,它不仅减少了实验中的不可控因素,同时还有分析速度快、消耗试剂少、设备简单、安全行好等优点。为提高度量结果的可信度来确保结果的适宜性。本文根据CNAS-GL006《化学分析中不确定度的评估指南》和HJ828-2017《水质挥发酚的测定流动注射-分光光度法》,对流动注射测定水中挥发酚的不确定度进行了评定[1-2]。
1 实验部分
1.1 仪器设备
全自动流动注射分析仪(北京吉天仪器FIA6000+FIA-6000+-01-1307088R),挥发酚分析通道(北京吉天仪器FIA6000+FIA-6000+-02(03)-1302080)。
1.2 药品
蒸馏试剂 (30%磷酸):将 150mL磷酸(H3PO4)(85%,优级纯)用超纯水稀释定容至500mL。
4-氨基安替比林显色剂:将0.32g4-氨基安替比林显色剂溶于水中,用纯水定容至500 mL。
铁氰化钾缓冲溶液(pH=10.3):将2.0g铁氰化钾[KFe(CN)3](优级纯),3.1g硼酸(H3BO3)(优级纯)和3.75g氯化钾(KCl)(分析纯)溶于900mL纯水中,加入1mol/L氢氧化钠(NaOH)溶液47mL,调节溶液的pH值为10.3,定容至1L。
载流:超纯水。
注:以上试剂除总氰化物蒸馏试剂外,所有试剂皆需超声30min脱气。
1.3 方法原理
用线蒸馏使挥发酚类化合物蒸馏,并与干扰物质和固定剂分离,被蒸馏出的酚类化合物,在pH10.0±0.2的介质中,在铁氰化钾的存在下,与4-氨基安替比林反应生成橙红色的安替比林燃料。在510nm进行比色测定[3]。
1.4 试验步骤
将分析仪挥发酚分析通道,自动进样器预热30min,安装蠕动泵管并连接好管路,打开流动注射软件工作站,配置仪器设置将挥发酚通道作为主通道,并连接自动进样器。设置测定参数(进样针清洗时间19s、达到阀时间290s、注射时间30s、进样时间140s、清洗时间60s、样品周期时间200s、出峰时间10s、峰宽40s),点击清洗程序设定清洗20min,待清洗完毕后,放好药剂,测试空白,稳定后开始正式测定[4]。
1.5 建立数学模型
流动注射软件采用最小二乘法对标准浓度和峰面积建立线性回归方程,方程为y=bx+a。其中y为样品峰面积;x为样品浓度mg/L;b为标准曲线斜率;a为标准曲线截距。
2 不确定度的评价
2.1 不确定度的来源分析
流动注射测定水中挥发酚实验过程中不确定度的来源:挥发酚标准溶液及稀释产生的不确定度;标准曲线引入的不确定度;样品重复检测的不确定度。
2.2 不确定度分量的量化
2.2.1 挥发酚标准溶液引入的不确定度
挥发酚标准溶液使用环保部标样所500mg/L标准样品,其相对扩展不确定度为%,呈正态分布,K=2,则标准储备液的标准不确定度分量为:

相对标准不确定度为:

2.2.2 由挥发酚标准储备液稀释成使用液引入的不确定度
挥发酚使用液采用逐级稀释,两次稀释10倍,一次稀释5倍。两次稀释10倍所用移液管和容量瓶规格一致,
1)由移液管转移溶液产生的不确定度。第一次稀释①校准。10mL移液管(A级)引入不确定度10mL移液管允许误差为±0.020mL,假设为三角分布,其相对不确定度为计算得到不确定度为。②温度。实验过程中温度变化为2℃,水体膨胀系数为2.1×10-4按照均匀分布计算其相对不确定度为 (10×2×以上两项合成不确定度为0.00085。两次相同方式稀释10倍不确定度为U。第三次稀释①校准。10mL移液管(A级)引入不确定度5mL移液管允许误差为±0.015mL,假设为三角分布,其相对不确定度为计算得到不确定度为0.015/(10×。②温度。实验过程中温度变化为2℃,水体膨胀系数为2.1×10-4按照均匀分布计算其相对不确定度为U(V3)=(10×2×2.1×10-4)。以上两项合成不确定度为。由三次转移稀释带来的不确定度为
由容量瓶转移溶液产生的不确定度:①校准,100 mL(A级)容量瓶最大允差为±0.10 mL,假设为三角分布,其相对不确定为0.010/(10×。②重复性,100 mL(A级)容量瓶刻度充满标准差为0.020 mL,其相对不确定为0.020/1000=0.00002。③温度,实验过程中温度变化为2℃,按均匀分布计算,其相对不确定度为。故单次过程引入的相对不确定度为U(V4)=三次稀释都使用相同规格100mL容量瓶,由三次稀释带来的相对不确定度为0.00048=0.00083。
综上,由挥发酚标准储备液及稀释成使用液引入的不确定度:
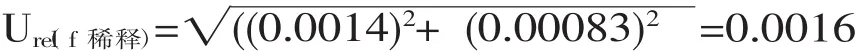
2.2.3 由标准曲线引入的不确定度
因使用线性最小二乘法拟合曲线程序的前提是假定横坐标的量的不确定度远小于纵坐标的量的不确定度,因此通常的C0不确定度计算程序仅仅与吸光度不确定度有关,而与校准曲线溶液的不确定度无关,也不与同一溶液中逐次稀释产生必然的相关性。顾认为标准溶液的不确定度足够小可以忽略,由标准曲线的不确定度由标准曲线拟合产生,标准曲线点为(0.0、2.0、5.0、10.0、20.0、50.0、100.0μg/L) 对每个标准点测量两次得到数据如下表1。
由以上数据使用最小二乘法拟合浓度-平均峰面积得到回归方程斜率b=0.0144,截距a=0.00584,残余标准差为:
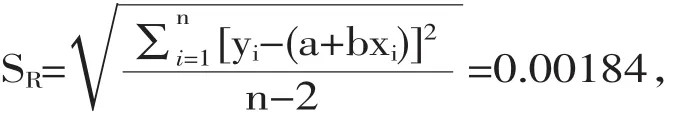

表1 标准曲线测定结果表
2.2.4 由重复测量样品引入的不确定度
重复测定挥发酚质控样200355(环标所,72.5±4.1ug/L) 7 次数据如表 2。

表2 质控样测定结果表
U(C0)==0.06,求得水样在标准曲线拟合下的相对不确定度为:
2.3 合成相对标准不确定度
综上所述,流动注射测定水中挥发酚合成相对标准不确定度为:

则合成标准不确定度为72.5×0.015=1.1μg/L,包含因子k=2,扩展不确定度为:Uc=k×uc=2×1.1=2.2μg/L
实验过程中各分量 Urel(c挥发酚)>Urel(m样品)>Urel(f稀释)>Urel(m拟合)。检测过程中不确定度主要来源于标准溶液、样品的重复性检测、标准溶液的稀释。
3 结论
1)根据计算流动注射测定水中挥发酚质控样的结果表示为 72.8±2.2 μg/L,k=2。
2)虽然在不确定度评定中没有计算标准曲线配置引入的不确定度,但是拟合曲线程序的前提是假定横坐标的量的不确定度远小于纵坐标的量的不确定度把它作为足够小的存在,这就要求标准曲线一定要严格按照规定配置使其符合相关要求。同时对重复测引入的不确定度所占分量较大,也许足够重视。
3)通过实验过程中不确定度的评估,可使分析人员更直观的了解到实验过程中哪些步骤带来的不确定度较高,在检测过程中予以重视,使最终的实验数据更可靠更科学。
猜你喜欢
中国应急管理科学(2021年4期)2021-04-13
化工设计通讯(2020年10期)2020-09-17
广西教育·C版(2020年4期)2020-07-09
中学生数理化·高一版(2020年9期)2020-01-02
中学化学(2019年4期)2019-08-06
考试周刊(2018年68期)2018-09-17
科教导刊(2017年26期)2017-11-07
中国氯碱(2016年9期)2016-11-16
读与写·下旬刊(2016年6期)2016-06-24
魅力中国(2016年12期)2016-02-05
