
利用Tn5转座子介导突变提高大肠杆菌丁醇生产水平
2015-06-05 09:35林兆董红军李寅
生物工程学报 2015年12期
林兆,董红军,李寅
利用Tn5转座子介导突变提高大肠杆菌丁醇生产水平
林兆1,2,董红军1,李寅1
1 中国科学院微生物研究所,北京 100101 2 中国科学院大学,北京 100049

林兆, 董红军, 李寅. 利用Tn5转座子介导突变提高大肠杆菌丁醇生产水平. 生物工程学报, 2015, 31(12): 1711–1719.Lin Z, Dong HJ, Li Y. Improvement of butanol production by Escherichia coli via Tn5 transposon mediated mutagenesis. Chin J Biotech, 2015, 31(12): 1711–1719.
目前,对于构建高产丁醇大肠杆菌工程菌株的工作,主要是对丁醇通路和相关途径的基因进行理性改造。为进一步提升菌株的丁醇生产能力,需要发掘基因组上可影响丁醇生产能力的基因,但这很难通过已有认识或计算机模型进行预测。本工作以一株实验室前期构建的产丁醇大肠杆菌工程菌株为研究对象,利用Tn5转座子构建了一个含有1 196个菌株的突变文库。丙酮酸是丁醇的前体,并且在发酵终产物中,副产物丙酮酸的含量与丁醇的含量呈反相关,因此,可以利用丙酮酸的含量来间接反映丁醇的含量,而丙酮酸可用二硝基苯肼显色法进行快速测定,基于此,建立了96孔板——酶标仪快速筛选方法。利用该方法成功筛选到了比对照菌株丁醇产量提高了29%、49%、56%的3个突变体菌株。利用反向PCR及测序的方法,确定了其转座子插入位置分别为:、、基因。这些基因可以作为进一步提高菌株丁醇产量的靶点,同时这种利用Tn5转座子筛选基因靶标的策略也为构建其他微生物细胞工厂提供了新思路。
大肠杆菌,丁醇,Tn5转座子,基因组
丁醇是一种重要的化学品,可直接用作有机溶剂和合成多种酯类化合物的前体,同时它也是一种比乙醇更有优势的生物燃料[1]。生物丁醇可由一些梭菌菌株在厌氧条件下经过ABE (Acetone-butanol-ethanol) 发酵产生,至今已有超过100年的历史[2]。但是由于梭菌菌株本身是严格厌氧的革兰氏阳性细菌,遗传操作困难,并且由于本身复杂的代谢调控机制,因此很难提升梭菌发酵生产丁醇的能力,从而限制了生物丁醇市场竞争力。借助于现代生物技术,科学家开始利用易于操作的模式细菌大肠杆菌来生产丁醇,取得了显著的进展。
2008年,来自University of California, Los Angeles的Liao研究组最先报道了把一条完整的梭菌丁醇发酵途径导入到大肠杆菌中(、、、、、、),第一次实现利用大肠杆菌生产丁醇[3]。在随后的几年里,大量的相关工作逐渐开展和发表,主要包括:1) 选择更为高效的酶。利用酶活更高的乙酰辅酶A乙酰转移酶AtoB (由大肠杆菌的基因编码) 来替代硫解酶Thl (由丙酮丁醇梭菌的基因编码),丁醇的产量提高了3倍[3];使用可催化不可逆反应且利用NADH为辅因子的顺式酰基辅酶A还原酶Ter (由) 替代丁酰辅酶A脱氢酶复合体BCD (由丙酮丁醇梭菌的、基因编码),使丁醇产量提高了17倍[4-5]。2) 还原力平衡设计。使用来自酵母菌的甲酸脱氢酶FDH (基因编码),可把厌氧发酵时产生的甲酸转化为二氧化碳和NADH,可实现丁醇合成的还原力平衡,在丰富培养基中丁醇得率最大可达36% (/)[4-5],也可以通过过表达丙酮酸脱氢酶PDH复合体(由基因编码),增强丙酮酸到乙酰辅酶A且偶联产生NADH的能力,丁醇得率可达到28% (/)[4]。3) 由于大肠杆菌本身存在一些副产物途径,从而影响了丁醇的产量和得率,去除这些副产物途径中关键基因(主要是产琥珀酸的、产乳酸的、产乙酸的和产乙醇的),可显著提高丁醇的合成能力[4-5],目前报道最高的大肠杆菌丁醇产量是14−15 g/L[5-6],略低于梭菌[7]。
基于理性知识指导的生产丁醇的大肠杆菌改造已经取得了显著的进展[8-9]。但是与梭菌相比较并没有绝对的优势,还远未达到有市场竞争力的产业化水平,仍需要更多的改进[10]。现在的菌株改造局限于有限的几个基因,然而细胞本身是一个系统性的整体,若要实现丁醇的高产,必然有基因组上其他基因的支持,但是具体哪些基因起到作用目前还难以预测。为了发现这些潜在的基因靶标,我们利用Tn5转座子[11-12]对一株可生产丁醇的大肠杆菌工程菌株进行了基因组水平的随机插入,构建了一个突变体文库,通过对文库筛选得到了丁醇产量提高的突变体,再对转座子插入位点进行定位,找出了有效的基因靶点,为构建更高效的产丁醇大肠杆菌提供了指导。
1 材料与方法
1.1 材料
1.1.1 菌株
实验所用菌株为大肠杆菌EB216菌株。EB216菌株是实验室前期根据Liao研究组[5]的工作构建,在15 mL BD小管中,利用M9培养基静置发酵能够产生1 g/L左右的丁醇。
1.1.2 试剂及引物
EZ-Tn5转座子试剂盒(EZ-Tn5™
二硝基苯肼购自国药集团化学试剂有限公司,溶于2 mol/L的HCl中制备0.1%的二硝基苯肼母液,最终稀释成0.037 5%的二硝基苯肼使用;NaOH购自北京化工厂,配成浓度为 2.81 mol/L溶液备用。
1.1.3 培养基
菌株发酵培养基为M9培养基,其1 L组成为:20 g葡萄糖,17.1 g Na2HPO4·12H2O,3.0 g KH2PO4,0.5 g NaCl,1.0 g NH4Cl,0.002 mol MgSO4,0.000 1 mol CaCl2,0.5 mg VB1,1 g酪蛋白氨基酸 (CA)。1 L的LB培养基组成为:10 g胰化蛋白胨,5 g酵母提取物,10 g NaCl。1 L的SOC培养基组成为:20 g胰化蛋白胨,5 g酵母提取物,0.5 g NaCl,2.5 mmol KCl,0.01 mol MgCl2,20 mmol葡萄糖。
pUC19筛选平板为含100mg/mL氨苄青霉素的LB琼脂平板,EZ-Tn5转座复合物转化子筛选平板为含50mg/mL卡那霉素的LB琼脂平板。
1.2 方法
1.2.1 感受态细胞制备至转化
EB216菌株的电转感受态细胞制备参考《分子克隆》[13]中的甘油法,制备感受态细胞使用pUC19质粒测试转化效率,以确保达到109/µg DNA以上。电击采用Gene Pulser Xcell电穿孔系统(Bio-Rad),使用2 mm电击杯,电击参数为:2 500 V,25 µF,200 Ω。
1.2.2 突变文库的制备
利用制备好的EB216感受态细胞,电转1 µLTn5转座子复合体。利用SOC液体培养基对电转菌株37 ℃复壮培养1 h后,涂布于含50mg/mL卡那霉素的LB琼脂平板。
1.2.3 用二硝基苯肼法测定丙酮酸
丙酮酸在酸性条件下与2,4-二硝基苯肼发生缩合反应形成丙酮酸二硝基苯腙。二硝基苯腙在碱性条件下呈现红色,可在520 nm处比色后灵敏地反映出丙酮酸的含量[14]。
以M9培养基作为溶剂,配置6.248 g/L的丙酮酸钠母液,再用M9培养基稀释成0、0.625、1.250、1.875、2.499、3.124、3.749、4.998 g/L的溶液,取80 µL不同稀释度丙酮酸钠溶液、80 µL 0.037 5%的二硝基苯肼、80 µL 2.81 mol/L NaOH混合,静置反应10 min,利用Molecular Device Spectra Max 190酶标仪在520 nm的波长下测定吸光度。每个梯度重复3次。
1.2.4 突变菌株96孔深孔板内发酵及筛选
将成功插入Tn5转座子的菌株,利用灭菌的牙签接种于96孔深孔板,每个孔装有1.25 mL的M9液体培养基,37 ℃条件下厌氧培养箱(Bactron I-2,Shellab) 中培养72 h。取发酵液200 µL于96孔酶标板,以空白为对照,利用酶标仪在600 nm的波长下测定吸光度。
同时将发酵液稀释25倍后利用二硝基苯肼法测定丙酮酸含量。
1.2.5 菌株的发酵评价
将最终筛选到的菌株进行小管发酵验证。首先在LB培养基里37 ℃培养过夜,转接0.5 mL的LB菌液至9.5 mL的M9培养基中,在厌氧培养箱中,利用15 mL BD试管(非密封) 于37 ℃厌氧条件静置培养72 h。取1.5 mL发酵液12 000×下离心1 min,将上清液过滤后利用HPLC (液相色谱仪) 检测丁醇的含量,以最终确定丁醇产量得到明显提高的菌株,对高产丁醇菌株进行3轮以上发酵重复验证,每轮3个平行,以证明其产量提高的稳定性。
1.2.6 利用HPLC对丁醇产量进行测定
丁醇产量利用高压液相(HPLC system 1260 Infinity Series, Agilent Technologies) 测定。所用分析柱为Bio-Rad (Hercules, CA) Aminex HPX-87H (7.8 mm×300 mm);流动相为5 mmol/L硫酸溶液,流速为0.5 mL/min,柱温是15 ℃;检测器为折光检测器,检测温度为30 ℃;检测时间为45 min。
1.2.7 反向PCR及测序确定Tn5插入位置
提取高产丁醇突变菌株的基因组DNA。根据EZ-Tn5结构,用限制性内切酶Ⅰ酶切基因组DNA。由于EZ-Tn5转座子上含有Ⅰ酶切位点,同时基因组上其他位置同样有Ⅰ位点,当细菌基因组被切成零碎片段后,用T4 DNA连接酶保证DNA片段自连接成环状DNA。以此为模板,以EZ-Tn5反向PCR引物(KAN-2 FP-1 Forward Primer及KAN-2 RP-1 Reverse Primer) 进行反向PCR,并用琼脂糖电泳分离纯化PCR产物。反向PCR产物通过北京擎科公司测序。测序引物同样为KAN-2 FP-1 Forward Primer及KAN-2 RP-1 Reverse Primer。根据测序结果来确定转座子插入位点。
2 结果与分析
2.1 构建Tn5转座子突变文库
根据试剂盒说明书,通过电击转化的方式将1 µL Tn5转座子复合体(20 ng) 导入自行制备的40 µL电转感受态细胞EB216中,可以得到100个左右的卡那霉素抗性菌落,这远低于说明书中给出的105个突变体/µL,这可能是由于菌株差异所致。通过转化10 µL转座子复合体,我们共计得到了1 196个抗性菌落文库,用于后续筛选。
2.2 建立高通量筛选方法
为了高通量筛选丁醇高产突变体菌株,需要建立有效的快速筛选方法。根据前期研究经验,我们发现作为丁醇前体的丙酮酸可以在胞外积累(浓度在0.5−1 g/L),且其浓度与丁醇浓度呈反相关,因此我们设想产丙酮酸少的突变体具有更高产丁醇能力,而丙酮酸可以通过二硝基苯肼显色法快速测定。基于此,我们配置了含不同浓度的丙酮酸溶液,将其与二硝基苯肼进行显色反应,利用酶标仪测定520 nm处吸光度(520) 时,发现酶标仪可直接测定0−100 mg/L浓度范围内的丙酮酸,并呈现出良好的线性关系,拟合线性回归方程为:=0.028 9,2=0.990 7。这也说明发酵液需要稀释才能直接测定,我们把发酵液稀释25倍后,用该法测出的发酵丙酮酸浓度与HPLC测定浓度基本一致,说明稀释后的菌体及发酵液其他成分不对丙酮酸测定形成干扰。
为了测定单位菌体产丙酮酸的能力,我们设计了如下筛选流程(图1):1) 突变体库接种至96孔深孔板培养,厌氧箱内培养3 d;2) 菌体吹吸均匀后,转移200 µL到96孔酶标板,测定菌体的光密度600值;3) 把菌液进行两次5倍稀释,得到25倍稀释液80 µL,先后加入80mL 0.037 5%的二硝基苯肼和80mL 2.81 mol/L NaOH进行显色反应,测定反应液的520值,根据丙酮酸标准曲线及稀释倍数算出发酵液中的丙酮酸浓度;4) 最后根据单位菌体的丙酮酸浓度(丙酮酸浓度/600) 筛选出突变体菌株,用于进一步分析丁醇生产能力。

图1 高产丁醇突变菌株筛选流程图
2.3 突变体筛选结果
利用上述方法对1 196个Tn5突变体进行了筛选,以丙酮酸浓度/600值评价菌株(图2),发现菌株分布呈现正偏态(朝左偏) 曲线,十分陡峭,大部分丙酮酸浓度/600值比较接近,没有发生显著变化,平均值为2.35 g/(L·600),我们选择低于平均值15%的200个菌株作为潜力菌株,通过小管发酵进一步筛选获得了16个菌株,经过了再次评价,筛选出了丁醇产量得到稳定提高的3个菌株进行后续评价。
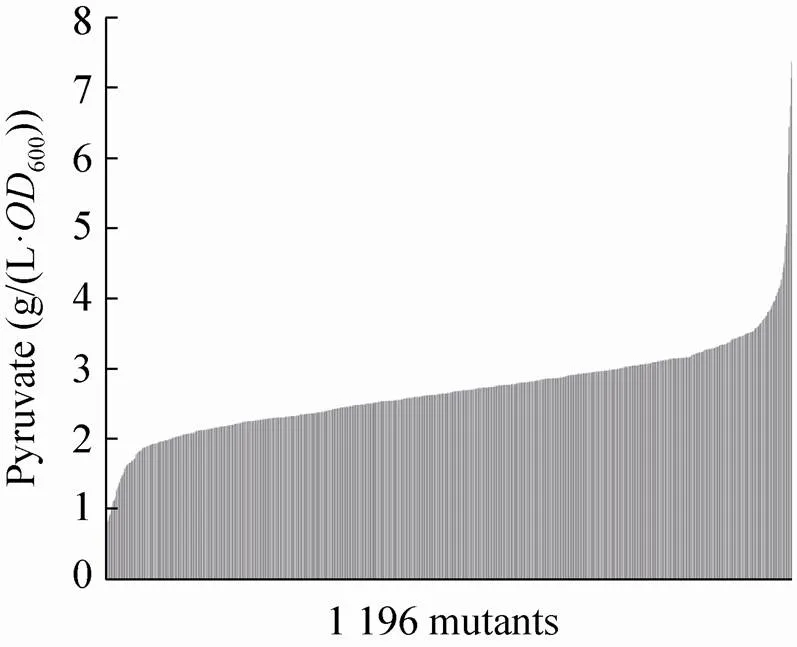
图2 突变菌株的丙酮酸浓度/OD600值
2.4 潜力菌株发酵能力的HPLC再分析
把上述筛选得到的菌株接种于含M9培养基的试管中进行发酵评价,用HPLC对发酵产物进行分析(表1),发现筛选得到的菌株丙酮酸含量比对照偏低,相应的丁醇产量则偏高,说明筛选策略是成功的。其中EB216-Tn5-17#、EB216-Tn5-304#、EB216-Tn5-599#丁醇产量分别提高了29%、49%、56%。
2.5 突变体菌株转座子插入位置的确定
通过对EB216-Tn5-17#、EB216-Tn5-304#、EB216-Tn5-599#三个菌株进行反向PCR,分别获得了1.8、2.8、1.2 kb左右的条带,经过测序,以及与大肠杆菌上的基因组进行对比,我们发现这3个菌株的插入位点分别为基因 (1 539 bp) 上的124/125位点、基因(618 bp)上的581/582位点以及基因(1 443 bp) 上的909/910位点(表2,图2)。
3 讨论
生物的生理代谢是一个复杂过程,由一个整体网络中的相互作用所决定。在菌株改造实践中,除了对已知的相关基因以及代谢通路进行改造外,在基因组上同样可能存在着一些目前我们不了解以及无法预测的、却对目的产物有影响的基因。我们利用Tn5转座子对一株产丁醇的大肠杆菌工程菌株构建了一个突变体文库,对其进行基于酶标仪的快速筛选,成功获得了丁醇产量提高的突变体,基于此获得的基因靶点为构建高产菌株提供了解决方案。
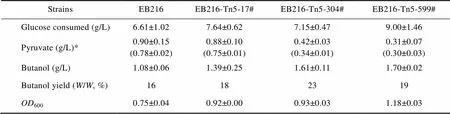
表1 突变菌株HPLC分析结果
*: the numbers in bracket are pyruvate concentrations detected by 2,4-dinitrophenylhydrazine reaction.
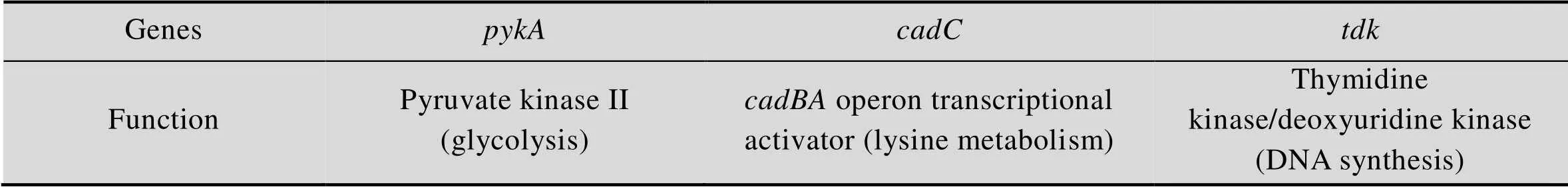
表2 转座子插入位点的鉴定
本研究中发现的3个基因中,基因编码胸苷激酶是DNA补救合成途径中的关键酶,又称为补救酶[15],它在Mg2+参与的情况下,催化胸苷磷酸化生成胸苷一磷酸,进而形成胸苷三磷酸,这是DNA合成中的4个必需脱氧核苷酸之一。胸苷激酶广泛存在于原核生物、真核生物以及少数病毒中[16]。已有研究发现,在经过驯化得到的生长速率加快的大肠杆菌中,和基因之间插入了一个IS片段,转录组分析发现该菌株中的转录水平显著下调[17]。这说明我们得到的失活突变体菌株,可能是由于生长速率的加快导致了丁醇产量的提高。是操纵子的转录活化子,处在的上游,负责激活编码的赖氨酸脱羧酶以及赖氨酸胺转运蛋白[18-19]。有研究表明在沙门鼠伤寒沙门氏菌中,如果敲除基因,糖酵解途径相关酶的活性会有所提高,同时突变菌株更能耐酸[20]。而在大肠杆菌的耐酸进化实验(pH 4.5−4.8) 中也发现,所有的进化种群都表现出赖氨酸脱羧酶活性的逐渐降低[21]。因此我们推测突变体中的糖酵解可能是加快了,从而利用了更多的葡萄糖生产丁醇,此外突变体的耐酸能力也有可能提升从而延长了丁醇的生产周期。基因编码丙酮酸激酶A,负责催化磷酸烯醇式丙酮酸(PEP) 形成丙酮酸以及ATP。它有一个丙酮酸激酶异构酶。通常认为是大肠杆菌中主要的丙酮酸激酶,而所起作用相对较小[22]。然而我们通过Tn5转座子插入到导致该基因的失活以及目的产物丁醇的提高,说明是一个参与了重要生理过程的基因,我们推测在厌氧条件下编码丙酮酸激酶是负责磷酸烯醇式丙酮酸转化为丙酮酸的关键酶,由于的失活,导致了生成的丙酮酸含量降低。细胞处在相对比较“饥饿”的状态,导致细胞更多地吸收外界的葡萄糖代谢以产生足够多的丙酮酸。同时这种“饥饿感”能够保证细胞更长时间处于“兴奋”状态,以致更迟进入衰退期,于是基因失活的菌株能够生成更多的丁醇。进一步生理及遗传学的研究,将可能会重新认识基因的生理功能,我们也正在进行该部分工作。
Tn5转座子插入突变是一种早就认识的遗传学机制,通常被用于发现特定功能的未知基因,主要应用于微生物遗传学研究[23]。我们将这种方法应用到菌株的遗传改造过程中,为高产菌株构建提供有效的靶点。相比于理性改造,它能够提供更多、更有效的靶点,相比于传统诱变,它没有大量的无效突变背景干扰,更容易确定关键基因,因此将是代谢工程的一种有效策略。为了发挥这种策略的最大功效,我们拟继续利用Tn5转座子进行更大规模的筛选,将利用自动接种仪进行高通量培养,利用自动移液工作站对发酵液进行显色测定,通过全基因组规模的饱和突变,找出所有可以提高丁醇产量基因靶点,然后整合这些靶点到一个菌株中,构建出高效的微生物细胞工厂。
[1] Green EM. Fermentative production of butanol-the industrial perspective. Curr Opin Biotechnol, 2011, 22(3): 337–343.
[2] Jones DT, Woods DR. Acetone-butanol fermentation revisited. Microbiol Rev, 1986, 50(4): 484–524.
[3] Atsumi S, Cann AF, Connor MR, et al. Metabolic engineering offor 1-butanol production. Metab Eng, 2008, 10(6): 305–311.
[4] Bond-Watts BB, Bellerose RJ, Chang MCY. Enzyme mechanism as a kinetic control element for designing synthetic biofuel pathways. Nat Chem Biol, 2011, 7(4): 222–227.
[5] Shen CR, Lan EI, Dekishima Y, et al. Driving forces enable high-titer anaerobic 1-butanol synthesis in. Appl Environ Microbiol, 2011, 77(9): 2905–2915.
[6] Dellomonaco C, Clomburg JM, Miller EN, et al. Engineered reversal of the β-oxidation cycle for the synthesis of fuels and chemicals. Nature, 2011, 476(7360): 355–359.
[7] Xue C, Zhao J, Zhao JB, et al. High-titer-butanol production byJB200 in fed-batch fermentation with intermittent gas stripping. Biotechnol Bioeng, 2012, 109(11): 2746–2756.
[8] Wen M, Bond-Watts BB, Chang MCY. Production of advanced biofuels in engineered. Curr Opin Chem Biol, 2013, 17(3): 472–479.
[9] Branduardi P, de Ferra F, Longo V, et al. Microbial-butanol production from Clostridia to non-Clostridial hosts. Eng Life Sci, 2014, 14(1): 16–26.
[10] Choi YJ, Lee JM, Jang YS, et al. Metabolic engineering of microorganisms for the production of higher alcohols. mBio, 2014, 5(5): e01524–14.
[11] Goryshin IY, Jendrisak J, Hoffman LM, et al. Insertional transposon mutagenesis by electroporation of released Tn5 transposition complexes. Nat Biotechnol, 2000, 18(1): 97–100.
[12] Reznikoff WS. Transposon Tn5. Annu Rev Genet, 2008, 42: 269–286.
[13] Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning: A Laboratory Manual. 2nd ed. New York: Cold Spring Harbor Laboratory Press, 1989: 119–123.
[14] Eze LC, Echetebu CO. Some properties of aspartate and alanine aminotransferases from trichodermaviride. J Gen Microbiol, 1980, 120(2): 523–527.
[15] Segura-Peña D, Lichter J, Trani M, et al. Quaternary structure change as a mechanism for the regulation of thymidine kinase 1-like enzymes. Structure, 2007, 15(12): 1555–1566.
[16] Sandrini MPB, Clausen AR, Munch-Petersen B, et al. Thymidine kinase diversity in bacteria. Nucleos Nucleot Nucl Acides, 2006, 25(9/11): 1153–1158.
[17] LaCroix RA, Sandberg TE, O'Brien EJ, et al. Use of adaptive laboratory evolution to discover key mutations enabling rapid growth of Escherichia coli K-12 MG1655 on glucose minimal medium. Appl Environ Microbiol, 2015, 81(1): 17–30.
[18] Küper C, Jung K. CadC-mediated activation of thepromoter in. J Mol Microbiol Biotechnol, 2005, 10(1): 26–39.
[19] Rhee JE, Kim KS, Choi SH. CadC activates pH-dependent expression of theoperon at a distance through direct binding to an upstream region. J Bacteriol, 2005, 187(22): 7870–7875.
[20] Lee YH, Kim BH, Kim JH, et al. CadC has a global translational effect during acid adaptation inserovar Typhimurium. J Bacteriol, 2007, 189(6): 2417–2425.
[21] Harden MM, He A, Creamer K, et al. Acid-adapted strains ofK-12 obtained by experimental evolution. Appl Environ Microbiol, 2015, 81(6): 1932–1941.
[22] Ponce E, Flores N, Martinez A, et al. Cloning of the two pyruvate kinase isoenzyme structural genes from: the relative roles of these enzymes in pyruvate biosynthesis. J Bacteriol, 1995, 177(19): 5719–5722.
[23] Miller WJ, Capy P. Mobile Genetic Elements. New Jersey: Humana Press, 2004: 83–96.
(本文责编郝丽芳)
Improvement of butanol production byvia Tn5 transposon mediated mutagenesis
Zhao Lin1,2, Hongjun Dong1, and Yin Li1
1 Institute of Microbiology, Chinese Academy of Sciences, Beijing 100101, China2 University of Chinese Academy of Sciences, Beijing 100049, China
For engineering an efficientbutanol-producingstrain,manyeffortshavebeenpaid ontheknowngenesorpathwaysbased on current knowledge. However, many genesinthegenomecould alsocontributetobutanol production in an unexpected way. In this work, we used Tn5 transposon to construct a mutant library including 1 196 strains in a previously engineered butanol-producingstrain. To screen the strains with improved titer of butanol production, we developedahigh-throughputmethodforpyruvatedetectionbased on dinitrophenylhydrazine reaction using 96-well microplatereader,becausepyruvateistheprecursorofbutanol anditsconcentrationisinverselycorrelatedwithbutanol in the fermentation broth.Usingthismethod, wesuccessfully screened three mutants with increased butanoltiter.Theinsertion sitesofTn5transposonwasintheORFsof,,andbyinversePCRandsequencing.Thesefound genes would be efficienttargetsforfurther strain improvement. And the genomescanning strategy described here will be helpful for other microbial cell factory construction.
, butanol, Tn5 transposon, genome
December 28, 2014; Accepted:January 22, 2015
Yin Li. Tel/Fax: +86-10-64807485; E-mail: yli@im.ac.cn
Hongjun Dong. Tel/Fax: +86-10-64807351; E-mail: redarmy305@gmail.com
10.13345/j.cjb.140646
Supported by:National High Technology Research and Development Program of China (863 Program) (No. 2011AA02A208), National Natural Science Foundation of China (No. 31270107).
国家高技术研究发展计划 (863计划) (No. 2011AA02A208),国家自然科学基金 (No. 31270107) 资助。
猜你喜欢
林业科学(2022年1期)2022-03-23
青岛科技大学学报(自然科学版)(2021年3期)2021-06-09
中国蜂业(2021年5期)2021-05-22
中国酿造(2020年10期)2020-11-04
分析仪器(2020年4期)2020-09-03
广西糖业(2016年1期)2016-12-19
浙江农林大学学报(2016年6期)2016-12-12
环境监控与预警(2016年5期)2016-04-18
环境监控与预警(2015年5期)2015-04-12
华东理工大学学报(自然科学版)(2014年5期)2014-02-27
