
利用合成的基因回路实现细胞水平上尿酸稳态控制的实验研究
2014-11-29 04:15曲国龙邵妤谭俊杰陈章金晶凌焱李玉霞刘刚陈惠鹏
生物技术通讯 2014年4期
曲国龙 ,邵妤,2,谭俊杰,陈章,金晶,4,凌焱,李玉霞,刘刚,陈惠鹏
1.军事医学科学院 生物工程研究所,北京 100071;2.安徽大学 生命科学学院,安徽 合肥 230039;3.成都军区总医院 呼吸科,四川 成都 610083;4.沈阳药科大学,辽宁 沈阳 110016
合成生物学(synthetic biology)是后基因组时代的新兴学科,同时也是现代生命科学研究的热点领域[1]。合成生物学新在“合成”二字,亦即一个再创造的过程,旨在以传统的生物学技术为基础,结合工程学、材料学、计算科学等其他学科的技术方法进行人工定向创造[2-3]。其涵盖的研究内容可大体分为3 个层次[4]:一是利用已知功能的天然生物模体(motif)或模块(module)构建新型调控网络并表现出新功能;二是采用从头合成(de novosynthesis)的方法,人工合成基因组DNA 并重构生命体;第三个层次则是在前两个研究领域得到充分发展之后,创建完整的全新生物系统乃至人工生命体(artificial life)。
近20 年来,合成生物学研究取得了长足进展。2000 年,《Science》发文报道了2 个人工合成的基因网络基因震荡器(oscillator)[5]和基因开关(toggles witch)[6];2002 年,Wimmer[7]研究组在历史上首次合成具有生物活性的脊髓灰质炎病毒基因组;2010 年5 月20 日,Venter[8]研究组通过将人工合成的支原体基因组移植到山羊支原体细胞,创造了首个仅由人造基因组控制的活细胞。这些具有里程碑意义的研究,充分体现了合成生物学广阔的前景及潜在的应用价值。比如,Keasling[9]等合成抗疟药物青蒿素和Voigt[10]等通过人工改造的细菌进行抗癌等经典研究工作,皆显示了合成生物学在生物医药领域的广泛应用。
尿酸(uric acid,UA)是嘌呤代谢的终产物,微溶于水,易形成尿酸盐晶体沉淀,长期的高尿酸血症易引发痛风。有文献报道,耐辐射异常球菌中,基因hucR编码的蛋白HucR 在尿酸介导下,可以与基因hucO产生相互作用[11-12],其作用原理是当尿酸浓度低于某一值时HucR 与hucO相结合,对hucO下游基因的表达产生抑制作用;当尿酸浓度高于某一值时,HucR 与hucO分离,hucO下游基因正常表达。2010年,有研究人员通过合成的基因回路实现了小鼠体内尿酸的稳态控制[13]。在本研究中,我们以mUTs(连有KRAB 结构域的hucR基因)和hucO8(hucO的8串联结构)为基础,分别构建双载体基因回路和双向共表达单载体基因回路,进一步研究分析细胞水平上回路对尿酸的感应及调控作用。
1 材料和方法
1.1 材料
HeLa 细胞、载体pcDNA3.1/V5-His(C)由本实验室保存;大肠杆菌DH5α化学感受态购自北京博迈德科技发展有限公司;载体pSEAP2-control 购自Clontech 公司;载体pBudCE4.1、转染试剂LipofectAMINE 2000和Amplex Red Uric Acid/Uricase Assay Kit 均购自Invitrogen 公司;dNTP、限制性内切酶、T4DNA 连接酶均购自NEB 公司;DNA marker购自Trans和TaKaRa公司;质粒小提试剂盒、DNA凝胶回收试剂盒购自Axygen 公司;LATaqDNA 聚合酶购自TaKaRa 公司;报告基因SEAP检测试剂盒购自TOYOBO 公司;DMEM 培养基、Opti-MEM 培养基、胎牛血清均购自GIBCO 公司;DMSO 购自Sigma 公司;胰蛋白酶购自HyClone 公司;24 孔和96 孔细胞培养板购自Costar 公司;尿酸干粉购自ACROS 公司;DNA 合成和测序均由北京奥科鼎盛生物科技有限公司完成。
1.2 mUTs及hucO8的合成
从NCBI 网站获 取mUTs及hucO8的序列,在mUTs的5'和3'端分别引入EcoRⅠ和NotⅠ酶切位点,hucO8的5'和3'端分别引入HindⅢ和EcoRⅠ酶切位点,人工合成2 段基因序列并连接至pMD18T 载体,分别命名为pMD18T-mUTs和pMD18T-hucO8。
1.3 mUTs基因的改造
通过PCR 在mUTs的5'端引入NotⅠ位点,3'端引入BglⅡ位点。用Primer Premier 5 设计PCR 上游引物P1(5'-GCGGCCGCGAATTCATGGATGCTA AGTCACTAA-3',下划线序列为NotⅠ位点)和下游引物P2(5'-AGATCTTCTAGATTATACCCCCTGCTC CAGCCC-3',下划线序列为BglⅡ位点)。PCR 模板为pMD18T-mUTs,PCR 扩增体系(25μL)包括DNA模板100 ng、LATaq(5 U/μL)酶0.3μL、引物(100μmol/L)各1μL、5×GC 缓冲液5μL、dNTP(2.5 mmol/L)4μL,用ddH2O 补至25μL。PCR 扩增程序:94℃预变性5 min,然后以94℃变性30 s、65℃退火1 min、72℃延伸1 min 扩增30 个循环,72℃延伸5 min。回收目的产物连接至pMD18T 载体,连接产物转化大肠杆菌DH5α感受态,挑取单菌落,振荡培养后提取质粒,酶切及测序鉴定,将阳性克隆命名为pT-NmUTsB。
1.4 载体pSEAP-hucO8的构建
以pMD18T-hucO8和pSEAP2-control 为基础载体,用HindⅢ/EcoRⅠ对载体分别进行双酶切,2%和1%的琼脂糖凝胶电泳分别分离回收pMD18T-hucO8小片段及pSEAP2-control 大片段,用T4DNA 连接酶连接2个目的片段,连接产物转化大肠杆菌DH5α感受态,挑取单菌落,培养后提取质粒,酶切鉴定。
1.5 转录抑制物表达载体pcDNA3.1/V5-mUTs的构建
以pMD18T-mUTs和pcDNA3.1/V5-His(C)为基础载体,用EcoRⅠ/NotⅠ对载体分别进行双酶切,1%琼脂糖凝胶电泳分离回收pMD18T-mUTs 小片段及pcDNA3.1/V5-His(C)大片段,用T4DNA 连接酶连接2个目的片段,连接产物转化大肠杆菌DH5α感受态,挑取单菌落,培养后提取质粒,酶切鉴定。
1.6 双向共表达载体pBudCE4.1-SEAP-mUTs 的构建
首先构建pBudCE4.1-SEAP。以pBudCE4.1和pSEAP-hucO8为基础载体,用HindⅢ/SalⅠ对载体分别进行双酶切,1%琼脂糖凝胶电泳分离回收pSEAP-hucO8小片段(hucO8-SEAP)及pBudCE4.1 大片段,用T4DNA 连接酶连接2 个目的片段,连接产物转化大肠杆菌DH5α感受态,挑取单菌落,培养后提取质粒,酶切鉴定。
以pBudCE4.1-SEAP和pT-NmUTsB 为基础载体,用NotⅠ/BglⅡ对载体分别进行双酶切,1%琼脂糖凝胶电泳分离回收pT-NmUTsB 小片段及pBudCE4.1-SEAP 大片段,用T4DNA 连接酶连接2个目的片段,连接产物转化大肠杆菌DH5α感受态,挑取单菌落,培养后提取质粒,酶切鉴定。
1.7 载体phucO8-smUox的构建
从NCBI 网站获取smUox序列,人工合成该基因序列并连接至pGH 载体,命名为pGH-smUox。以pSEAP-hucO8和pGH-smUox 为基础载体,用EcoRⅠ/XbaⅠ对载体分别进行双酶切,1%琼脂糖凝胶电泳分离回收pGH-smUox 小片段及pSEAP-hucO8大片段,用T4DNA 连接酶连接2 个目的片段,连接产物转化大肠杆菌DH5α感受态,挑取单菌落,培养后提取质粒,酶切鉴定。
1.8 单载体UA 调控回路pBudCE4.1-mUTs-smUox的构建
首先构建载体pBudCE4.1-smUox。以载体phucO8-smUox和pBudCE4.1 为基础载体,用Hind Ⅲ/BamHⅠ对载体分别进行双酶切,1%琼脂糖凝胶电泳分离回收phucO8-smUox 小片段(hucO8-smUox)及pBudCE4.1 大片段,用T4DNA 连接酶连接2 个目的片段,连接产物转化大肠杆菌DH5α感受态,挑取单菌落,培养后提取质粒,酶切鉴定。
然后以pBudCE4.1-smUox和pBudCE4.1-SEAP-mUTs为基础载体,用NotⅠ/BglⅡ对载体分别进行双酶切,1%琼脂糖凝胶电泳分离回收pBudCE4.1-SEAP-mUTs 小片段及pBudCE4.1-smUox 大片段,用T4DNA 连接酶连接2 个目的片段,连接产物转化大肠杆菌DH5α感受态,挑取单菌落,培养后提取质粒,酶切鉴定。
1.9 HeLa细胞培养及转染
HeLa 细胞用含10%胎牛血清的DMEM 培养基在37℃、5% CO2的培养箱中培养。细胞复苏后传代2~3次,至细胞状态良好时进行转染;转染前24 h接种HeLa 细胞到24 孔细胞培养板中,每孔约1.0×105细胞,以保证转染时细胞融合度达到90%左右。转染步骤详见LipofectAMINE 2000说明书。
1.10 报告基因SEAP检测
用Reporter Assay Kit(-SEAP-)在Enspire Multilabel Reader(Perkin Elmer)上检测分泌型碱性磷酸酶(secreted alkaline phosphatase,SEAP)的表达量。瞬时转染检测时间为24~48 h,SEAP 的表达是一个持续的过程,48 h 后表达量相对较大,检测结果相对较好。具体步骤详见试剂盒说明书。
1.11 20 mmol/L尿酸溶液的配制
配制100 mL 50 mmol/L(pH10.0)的Tris-HCl溶液,称取336 mg UA 粉末,倒入Tris-HCl 溶液中,搅拌至UA粉末完全溶解,用0.22μm的一次性过滤器过滤后用于细胞转染实验。
1.12 尿酸及尿酸酶检测
转染后48 h,用Amplex Red Uric Acid/Uricase Assay Kit 在Enspire Multilabel Reader 上检测尿酸及尿酸酶含量,检测波长560 nm。具体步骤详见试剂盒说明书。
2 结果
2.1 载体pSEAP-hucO8和pcDNA3.1/V5-mUTs 的构建
挑取构建后载体的单克隆,用HindⅢ/EcoRⅠ双酶切,2%琼脂糖凝胶电泳分离酶切产物,结果如图1A,250 bp 偏上条带与目的片段(274 bp)相符,说明载体pSEAP-hucO8构建成功。
挑取构建后载体的单克隆,用EcoRⅠ/NotⅠ双酶切,1%琼脂糖凝胶电泳分离酶切产物,结果如图1B,1000 bp 处出现条带,与目的片段(974 bp)相符,说明载体pcDNA3.1/V5-mUTs构建成功。
2.2 载体pBudCE4.1-SEAP-mUTs的构建
1%琼脂糖凝胶电泳鉴定PCR 产物,结果如图2A,1000 bp 处出现目的条带;目的条带连接至pMD18T 载体,用NotⅠ/BglⅡ双酶切,电泳结果如图2B,部分样品出现目的条带,测序分析出现目的条带的单克隆样品,结果正确,说明在mUTs的5'和3'端成功引入NotⅠ和BglⅡ位点。挑取构建的pBudCE4.1-SEAP 单克隆,用HindⅢ/SalⅠ双酶切,1%琼脂糖凝胶电泳结果如图2C,2000 bp 偏上出现条带,与目的片段(2301 bp)相符,说明载体pBudCE4.1-SEAP 构建成功。挑取构建的pBudCE4.1-SEAP-mUTs 单克隆,分别用HindⅢ/SalⅠ和NotⅠ/BglⅡ双酶切,1%琼脂糖凝胶电泳结果如图2D,3 个单克隆样品均出现与2 个目的片段大小相符的条带,说明载体pBudCE4.1-SEAP-mUTs构建成功。
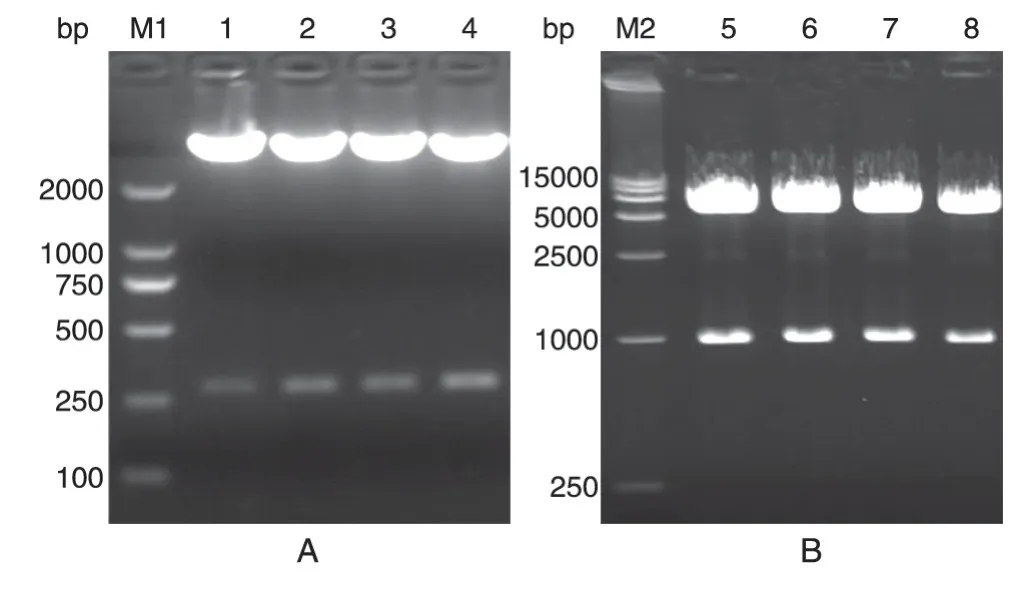
图1 琼脂糖凝胶电泳鉴定pSEAP-hucO8(A)和
2.3 单载体UA 调控回路pBudCE4.1-mUTs-smUox的构建
挑取phucO8-smUox 载体单克隆,用EcoRⅠ/XbaⅠ双酶切,1%琼脂糖凝胶电泳结果如图3A,1000 bp 处出现条带,与目的片段(985 bp)相符,说明载体phucO8-smUox 构建成功。挑取pBudCE4.1-smUox 载体单克隆,用HindⅢ/BamHⅠ双酶切,结果如图3B,1500~2000 bp 间出现条带,与目的片段(1755 bp)相符,说明pBudCE4.1-smUox 构建成功。以NotⅠ/BglⅡ为酶切位点,在pBudCE4.1-smUox 载体上连入mUTs,完成载体pBudCE4.1-mUTs-smUox的构建。用HindⅢ/BamHⅠ和NotⅠ/BglⅡ分别进行双酶切,结果如图3C,出现2 个与目的片段(985、1755bp)相符的条带,且大片段正确,说明载体pBudCE4.1-mUTs-smUox构建成功。
2.4 双载体基因回路基本功能验证
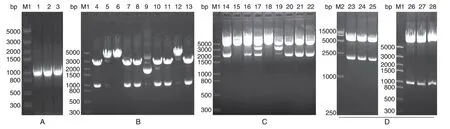
图2 载体pBudCE4.1-SEAP-mUTs构建的琼脂糖凝胶电泳鉴定结果
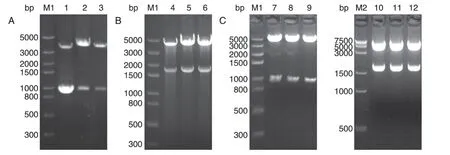
图3 载体pBudCE4.1-mUTs-smUox构建的琼脂糖凝胶电泳鉴定
以不同浓度LipofectAMINE 2000 转染pSEAPhucO8(每孔0.8μg)至HeLa 细胞,转染48 h 后收集细胞培养基上清进行SEAP化学发光强度检测,结果如图4A,随着LipofectAMINE 2000 浓度的提高,SEAP 的表达量增加,说明pSEAP-hucO8转入细胞后SEAP基因可以正常表达。以不同摩尔比共转染pcDNA3.1/V5-mUTs和pSEAP-hucO8(固定每孔0.8μg),2 种质粒与LipofectAMINE 2000 分别稀释、混合,转染时每孔各加入100μL 混合液,转染6 h 后换液,转染48 h后检测,结果如图4B,相比于单独转染pSEAP-hucO8,共转染pcDNA3.1/V5-mUTs和pSEAP-hucO8时,SEAP 化学发光强度显著降低,且随着mUTs/hucO8比例的提高,SEAP发光强度逐渐缓慢降低。说明mUTs可以正常表达且能与hucO8结合并抑制下游基因表达,随着mUTs/hucO8摩尔比的提高,抑制作用增强。
2.5 单载体回路pBudCE4.1-SEAP-mUTs 基本功能验证
以不同浓度的LipofectAMINE 2000 转染pBudCE4.1-SEAP(每孔1.0μg)至HeLa 细胞,转染48 h 后收集细胞培养基上清进行SEAP 化学发光强度检测,结果如图5A,随着LipofectAMINE 2000 浓度的提高,SEAP 化学发光强度增强,说明pBudCE4.1-SEAP 转入细胞后SEAP基因可以正常表达;然后分别以不同摩尔比转染pBudCE4.1-SEAP-mUTs(每孔1.2μg)和pcDNA3.1/V5-mUTs,转染48 h后检测SEAP化学发光强度,结果如图5B,实验组相比于阴性对照组SEAP 化学发光强度明显降低,且mUTs/hucO8比例越高,发光强度越低。说明pBudCE4.1-SEAP-mUTs 载体上mUTs可以正常表达且能与hucO8结合并对下游基因表达起到一定的抑制作用,且mUTs/hucO8比例越高,抑制作用越明显。cO8/mUTs=1∶4。
2.6 回路对尿酸感应作用分析
通过检测不同浓度尿酸培养时SEAP 的表达量来验证回路对尿酸的感应作用。以4∶1 的比例共转染pcDNA3.1/V5-mUTs和pSEAP-hucO8或者单独转染pBudCE4.1-SEAP-mUTs,转染6 h 后换液并添加不同浓度的UA,转染48 h 后检测SEAP 化学发光强度。共转染结果如图6A,0~0.3 mmol/L 范围内SEAP 化学发光强度基本不变,0.4~2.0 mmol/L 范围内SEAP 化学发光强度明显增强,但变化不大;单独转染pBudCE4.1-SEAP-mUTs 结果如图6B,0~2.0 mmol/L 范围内SEAP 化学发光强度逐渐增强,且变化明显。结果表明,双载体回路和单载体回路均具有一定的尿酸感应能力,单载体回路在0~0.3 mmol/L 尿酸浓度范围内感应作用不明显,0.4~2.0mmol/L范围内感应作用明显,但变化不大;单载体回路在0~2.0 mmol/L 范围内对UA 的感应作用逐渐增强且变化明显。

图4 双载体基因回路基本功能验证(SEAP化学发光强度检测)
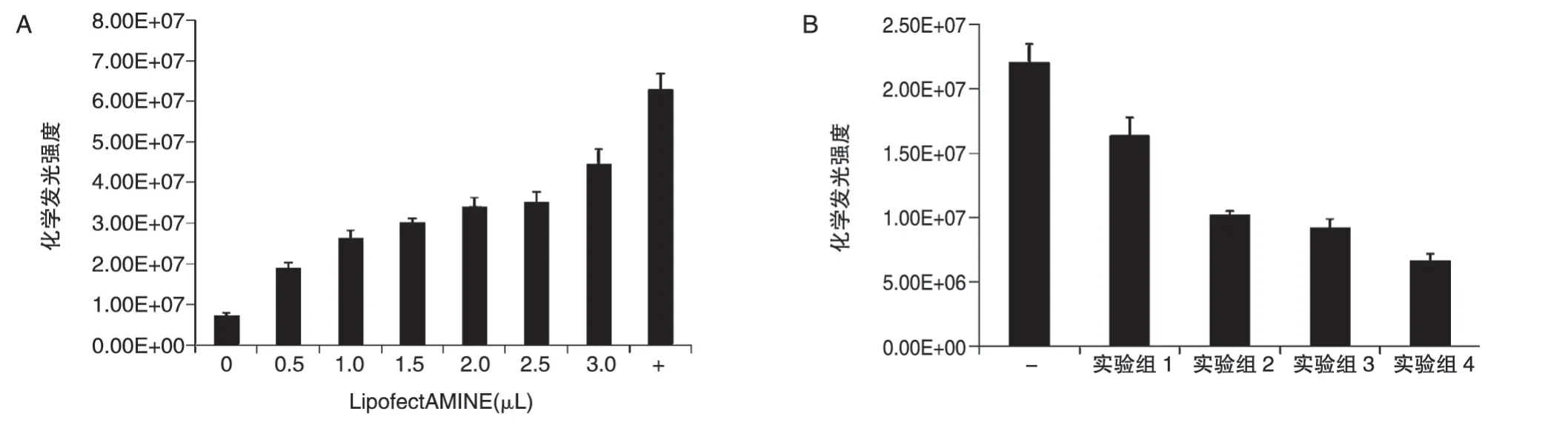
图5 单载体回路pBudCE4.1-SEAP-mUTs基本功能验证(SEAP化学发光强度检测)
2.7 回路对尿酸调控作用分析
单独转染pBudCE4.1-smUox,检测smUox能否在HeLa 细胞中正常表达,转染48 h 后检测培养基中尿酸酶的含量,结果如图7A,随着pBudCE4.1-smUox 浓度的提高,培养基中尿酸酶含量逐渐增加。设计不同的分组实验,检测回路对尿酸的调控作用,转染6 h 后换液,每孔加入500μmol/L 尿酸,转染48 h后检测培养基中的UA含量,结果如图7B,与阴性对照相比,实验组培养基中UA浓度均有所下降。最后以3∶1 的摩尔比共转染pcDNA3.1/V5-mUTs和pBudCE4.1-mUTs-smUox,转染6 h后换液,分别添加不同浓度的UA,转染48 h 后检测培养基中的UA 浓度,以此检验回路对UA 的调控能力及调控范围,结果如图7C,0~300μmol/L 范围内尿酸浓度基本不变,500~2000μmol/L 范围内尿酸浓度均有所下降,且初始浓度越高,下降幅度越大。综合分析表明,smUox转染HeLa 细胞后可以正常表达并具有活性;单载体回路及双载体回路均具有一定的尿酸调控能力,且尿酸调控能力相当;当mUTs/hucO8=4∶1 时,回路对尿酸的调控作用略有增强,但变化不大;尿酸浓度为0~2000μmol/L,pcDNA3.1/V5-mUTs 与pBudCE4.1-mUTs-smUox 的摩尔比为3∶1时,0~300μmol/L 范围内回路对尿酸基本没有调控作用,500~2000μmol/L 范围内,随着尿酸浓度增高,回路的降尿酸作用相对越强。
3 讨论
合成生物学作为我国亟待发展的前沿科学正逐步被了解和重视,虽然目前还处于起步阶段,但从现有研究成果不难看出,其在生物医药、能源、环境污染治理、生物传感器和军事等诸多领域具有极好的应用前景。利用已知功能的基因片段,人工合成一些新的基因装置或调控网络,从而定向改变哺乳动物细胞的代谢行为,是合成生物学在生物医药领域的一个研究热点。
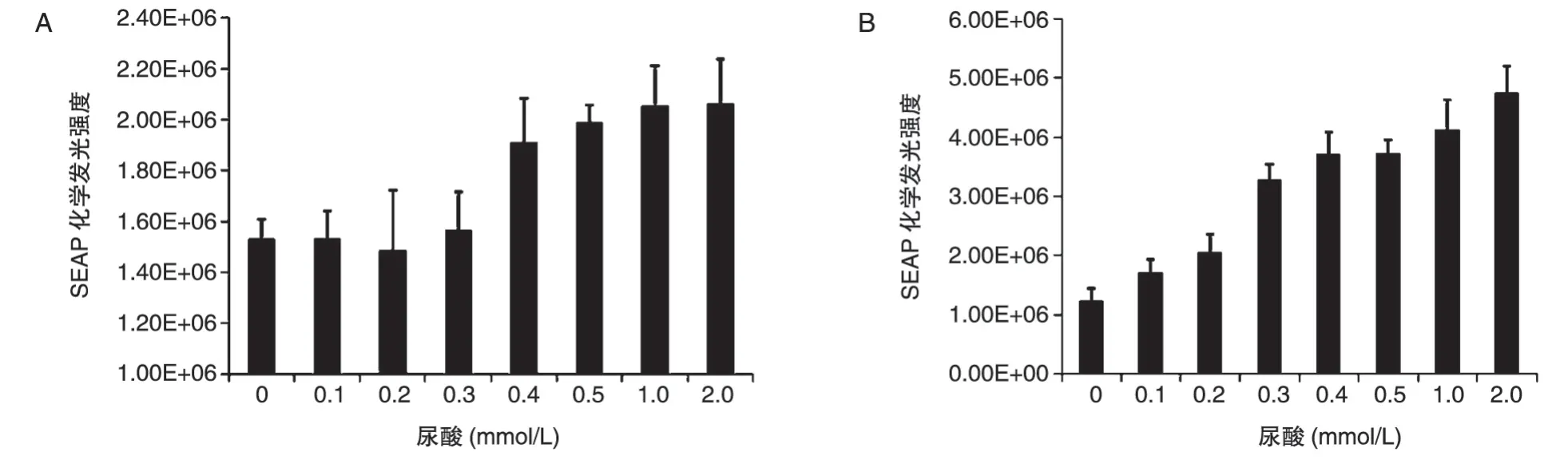
图6 回路对尿酸感应作用分析
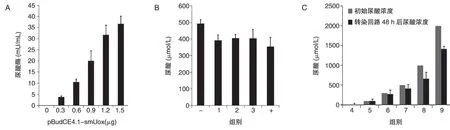
图7 回路对尿酸调控作用分析
本研究中,我们合成的基因回路就是利用了耐辐射异常球菌基因组中已知功能的基因hucR及其结合位点hucO。hucR经过起始密码子优化,连接蛋白结构域形成优化的转录抑制物基因mUTs,hucO经过串联形成结合位点的8串联结构hucO8,再以mUTs和hucO8为基础,利用合成生物学的方法,定向构建新的尿酸调控回路。回路发挥功能的前提是:mUTs转录翻译后产生的蛋白mUTS 具有转录抑制作用,且mUTS和hucO8在尿酸的介导下可以相互结合或分离。回路的基本原理是:当尿酸低于某一浓度时,mUTS 与hucO8结合,抑制hucO8下游基因的表达;当尿酸高于某一浓度时,mUTS 与hucO8分离,hucO8下游基因正常表达。尿酸酶具有分解尿酸的作用,当我们把尿酸酶基因连入hucO8下游时,回路就可以对周围环境中不同浓度的尿酸做出相应的应答。当尿酸低于某一浓度时,回路关闭,不发挥调控作用;当尿酸浓度高于某一浓度时,hucO8下游的尿酸酶基因正常表达,从而降低尿酸浓度,当尿酸浓度降低至某一值时,mUTS与hucO8相结合,抑制尿酸酶基因的表达,使尿酸浓度稳定在一定范围内,从而实现对尿酸的调控。
我们首先利用优化的基因片段mUTs和hucO8完成了载体pcDNA3.1/V5-mUTs和pSEAP-hucO8的构建,组成了双载体基因回路,通过HeLa 细胞转染实验,验证了mUTs和hucO8在尿酸介导下的相互作用;然后利用基因片段mUTs和hucO8合成了双向共表达载体pBudCE4.1-SEAP-mUTs,实现了单载体基因回路的构建,通过细胞转染实验,验证了单载体回路上mUTs和hucO8在尿酸介导下的相互作用;最后通过连入优化的黄曲霉菌尿酸酶基因smUox,构建了单载体尿酸调控回路pBudCE4.1-mUTs-smUox,通过检测回路转染HeLa 细胞后培养基中尿酸浓度的变化,验证了回路对尿酸的调控作用。我们发现,mUTs与hucO8的比例对回路影响很大,当二者比例为4∶1 时,mUTs对下游基因表达的抑制作用明显增强,从而影响回路对尿酸的调节范围及调节程度。鉴于LipofectAMINE 2000 的细胞毒性、转染效率等因素,我们没有继续增大二者比例进行实验研究。
相比于双载体基因回路,单载体基因回路具有以下特点:单载体基因回路可以规避转染效率不定导致的转入细胞的mUTs和hucO8比例不定的问题,使得mUTs与hucO8可以按照1∶1 的比例转入细胞;相比于通过共转染才能实现的基因回路,单载体回路在一定程度上降低了实验操作的难度;单载体回路在很大程度上简化了后续研究中稳定转染细胞系的筛选工作。
我们构建的基因回路也存在一些问题,与预期结果有一些差异。其中比较主要的问题是,单载体和双载体基因回路对尿酸都具有一定的感应调节作用,但调节作用不够理想。比如,尿酸浓度为1000~2000μmol/L时,虽然基因回路的降尿酸程度相对较大,但仍不能降至正常生理范围内。我们虽然发现当mUTs与hucO8比例为4∶1 时对回路会有较大影响,但由于技术上的原因,未能成功构建二者比例为4∶1 的单载体回路,为了达到更好的效果,仍须共转染pcDNA3.1/V5-mUTs 来提高mUTs的比例。我们通过瞬时转染的方法验证了回路对尿酸具有一定的感应及调节能力,但由于没有进行稳定转染细胞系的筛选,所以无法对尿酸浓度不断变化时,回路是否具有可逆性及回路的长期稳定性进行验证。这些存在的问题仍需要在后续研究中进一步探索解决。
正是合成生物学的兴起与发展,才使得我们可以利用已知功能的生物模块,经过简单的设计,合成新型调控网络并使其表现出预期的功能。这种研究方法为一些代谢类疾病的基因及细胞水平上的治疗提供了新的思路。相信随着合成生物学技术的进一步发展,以合成生物学为基础的相关研究会逐步走向临床应用、走进生活,在实践中发挥更大的作用。
[1]楼铁柱.合成生物学发展回顾与军事应用前景展望[J].军事医学,2011,35(2):81-87.
[2]Kanehisam M.Post-genome informatics[M].New York:Oxford University Press,2000:148.
[3]赵心清,姜如娇,白凤武.启动子和细胞全局转录机制的定向进化在微生物代谢工程中的应用[J].生物工程学报,2009,25(9):1312-1315.
[4]凌焱,段海清,陈惠鹏.合成生物学[J].军事医学科学院院刊,2006,30(6):572-574.
[5]Elowitz M B,Leibler S.A synthetic oscillatory network of Transcriptional regulators[J].Nature,2000,403(6767):335-338.
[6]Gardner T S,Cantor C R,Collins J J.Construction of a genetic toggle switch in Escherichia coli[J].Nature,2000,403(6767):339-342.
[7]Cello J,Paul A V,Wimmer E.Chemical synthesis of poliovirus cDNA:generation of infectious virus in the absence of natural template[J].Science,2002,297(5883):1016-1018.
[8]Gibson D G,Glass J I,Lartigue C,et al.Creation of a bacterial cell controlled by a chemically synthesized genome[J].Science,2010,329(5987):52-56.
[9]Ro D K,Paradise E M,Ouellet M,et al.Production of the antimalarial drug precursor artemisinic acid in engineered yeast[J].Nature,2006,440(7086):940-943.
[10]Levskaya A,Chevalier A A,Tabor J J,et al.Synthetic biology:engineering Escherichia coli to see light[J].Nature,2005,438(7067):441-442.
[11]Wilkinson S P,Grove A.HucR,a novel uric acid-responsive member of the MarR family of transcriptional regulators from Deinococcus radiodurans[J].J Biol Chem,2004,279:51442-51450.
[12]Wilkinson S P,Grove A.Negative cooperativity of uric acid binding to the transcriptional regulator HucR from Deinococcus radiodurans[J].J Mol Biol,2005,350:617-630.
[13]Kemmer C,Gitzinger M,Daoud-EI Baba M,et al.Self-sufficient control of urate homeostasis in mice by a synthetic circuit[J].Nat Biotechnol,2010,28:355-360.
猜你喜欢
太原理工大学学报(2021年6期)2021-11-25
中国海洋大学学报(自然科学版)(2019年7期)2019-05-21
中国海洋大学学报(自然科学版)(2019年7期)2019-01-04
科技视界(2018年19期)2018-10-09
山西医科大学学报(2017年11期)2017-12-01
广州化工(2016年11期)2016-09-02
天津科技大学学报(2016年3期)2016-08-02
哈尔滨商业大学学报(自然科学版)(2016年1期)2016-04-22
安徽理工大学学报·自然科学版(2015年1期)2015-07-21
