
论医学研究知情同意的伦理审查
2014-05-25 04:39胡晋红
中国医学伦理学 2014年2期
胡晋红
(第二军医大学长海医院药学部,上海 200433,hjhong2016@126.com)
1 知情同意的定义
知情同意[1]指向受试者告知一项试验的各方面情况后,受试者自愿确认其同意参加该项临床试验的过程,须以签名和注明日期的知情同意书作为文件证明。
2 医学研究知情同意获得的相关规定
2.1 知情同意的过程
1996年,美国、欧洲、日本医药管理机构联席召开的国际会议通过的《临床实践指南》明确规定:“知情同意是一个过程,个人通过这个过程在了解了决定参与试验的所有相关方面之后,自愿表达他或她参加该项试验的意愿”。美国生命伦理顾问委员会特别强调知情同意是获得同意的动态过程,而不是用书面、签字等形式获得文书的过程,不能允许文件本身成为过程。中国食品药品监督管理总局发布的《药物临床试验质量管理规范》、《医疗器械临床试验质量管理规范(征求意见稿)》也规定在受试者参与临床试验前,研究者应当充分向受试者或其家属、监护人、法定代理人说明临床试验中已知的、可预见的风险和可能发生的不良事件等详细情况,经充分和详细解释后由受试者或其法定代理人在《知情同意书》上签署姓名和日期,执行知情同意的研究者也需在知情同意书上签署姓名、注明日期。可见:知情同意不能仅凭一纸表格来证明和取代知情同意的过程。检查、判断一个项目是否做到了知情同意,不能仅仅看参与者的签字,而要看参与者是否充分了解研究者所属的机构、研究目的、过程、方法、资金的来源、预期的受益、可能的利益冲突、潜在的风险和研究可能引起的不适;要看他们的同意是否是在充分知情而且完全不受胁迫利诱的情况下作出的,还要看他们是否了解自己在参与医学研究过程中的权益。
2.2 获得知情同意的原则
知情同意原则包含信息、理解和自愿3个要素。在获得知情同意的过程中应注意遵循完全告知、充分理解、自主选择的原则;对如何获得知情同意或授权同意有详细说明并及时告知受试者签署知情同意书的相关规定和注意事项;知情同意所用语言适合该受试者群体理解;知情同意应明确表述在获得新的与研究风险相关的信息时需要再一次签署;知情同意的获得应在受试者有足够时间阅读信息、咨询研究者及考虑成熟后决定。
3 伦理委员会对医学研究知情同意的审查
3.1 伦理委员会对医学研究知情同意书中告知信息完整性的审查
《药物临床试验伦理审查工作指导原则》、《医疗器械临床试验质量管理规范(征求意见稿)》均规定了研究者或其指定的代表必须向受试者说明有关临床试验的详细情况,并列出了知情同意书中告知信息应涵盖的内容(见表 1)。[2-3]

表1 医学研究知情同意书告知信息涵盖的内容
伦理委员会对医学研究知情同意书中告知信息审查时,要按以上相关规定逐项对照是否将研究内容、试验风险、预期受益、隐私保密及医疗保护、损害赔偿等信息充分和完整的告知了受试者。目前有些知情同意书告知信息有疏漏或不完整:对试验风险叙述轻描淡写;对阳性对照药或联合用药可能发生的风险告知遗漏或过于简单;对设置安慰剂对照组的医学研究不明确告知受试者可能进入安慰剂组;对受试者可以自由选择本研究治疗方法外的其他有效治疗方法的信息未告知;对试验过程中取血的频度和总量、访视的次数和间隔等告知不具体;对受试者“无需任何理由,可随时退出试验”及“不会受到歧视和报复”的条文遗漏或强调不够;特别是对受试者如发生与其参与的医学研究相关的损伤后应获得的“赔偿”原则有意回避或含糊其辞;对出现紧急情况时受试者需接洽的联系人和联系办法不明确等。对这些知情同意书中容易出现的问题,伦理委员会审查时都应该加以关注和指出告知不充分之处,并要求研究者加以修正。
3.2 伦理委员会对医学研究知情同意书签署过程的审查
知情同意书须由受试者或其法定代理人在知情同意书上签字并注明日期,执行知情同意过程的研究者也需在知情同意书上签署姓名、注明日期。按照《药物临床试验伦理审查工作指导原则》及《医疗器械临床试验质量管理规范(征求意见稿)》的相关规定,具体知情同意书签署过程可按图1执行。
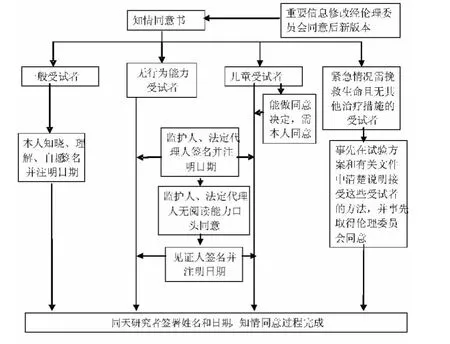
图1 知情同意过程
伦理委员会对医学研究知情同意书签署过程审查时要关注是否按要求实施知情同意书的签署。目前知情同意书受试者/法定代理人的签署不符合规范的常见问题有:①知情同意书是由研究者或其他人代替受试者签署,在医学研究项目质量控制检查中询问受试者时,有的受试者对知情同意书内容完全不知晓;②研究者代替受试者在知情同意书上签署日期;③对无行为能力受试者没有按要求由法定代理人签署知情同意书;④签署知情同意书的父母(早婚)没有达到法定年龄。在知情同意实施过程中存在的问题主要有:①再次获得知情同意的过程不规范,当发现对受试者有风险增加的重要信息时,并没有及时告知受试者,而是拖延数月之久才再次签署新版知情同意书;②由于需要对知情同意书进行了修改,但部分受试者没有签署新的修正后的知情同意书,仍签署的是旧版知情同意书;③知情同意过程中没有征得能做同意决定的大龄儿童受试者的同意。因此在伦理跟踪审查中需密切关注以上问题并加以纠正,同时增强培训。
3.3 伦理委员会对医学研究知情同意书用语规范性的审查
受试者及其亲属具有一定的理解和自主决定能力,医务人员具有丰富的专业知识、医学研究经验和高尚的医德修养,所有医学研究的参与者在病人利益和相互价值观念上能达成一致,是知情同意原则实现的必要条件。因此知情同意书在实施中特别要注意:①告知清楚、用语规范、简明易懂;② 不推卸应承担的责任;③不诱导受试者参与试验;④不放弃受试者应有的权益。目前知情同意实施中常会碰到一些问题,如某知情同意书中额外说明“此研究为你提供了良好的医疗服务”,本意是好的,但却容易让人误解研究之外不能得到良好的服务;又如某知情同意书中为了说明研究的针对性,使用“用药到病情恶化”、”要进入此试验必须得癌症”等非常不适的词语。还有的知情同意书使用了“诊断检查的此项优惠可为你节省10000元的治疗费用”等具有诱导性的表述;甚至有知情同意书还有“本产品的使用风险都已得到了控制”、“此研究方案为顶级专家设计”、“本研究产品安全性更高”、“不会导致不良反应,或其他并发症”等不恰当表述;而“由于参加此研究发生与试验相关的非医疗疏漏造成的损伤时,将由申办方负责提供救治费用和相应赔偿”、“您可选择参加本试验,也可选择其他的临床常规治疗措施如……”等涉及受试者权益的词句在有些知情同意书中缺失。伦理委员会在审查知情同意书时应对不规范的表述、过于专业化及晦涩难懂不利于理解的用语以及由于翻译效果不符合中国用语习惯易引起误解的表述提出修正;对具有诱导性的语言要求删除;对缺乏体现受试者权益保护的内容提出增补。
3.4 伦理委员会对医学研究知情同意书风险处置的审查
伦理委员会在审查知情同意时应确认知情同意书已清楚告知了受试者与其联系的方式、隐私及保密条款,提供医疗、社会支持的措施,因研究所致伤害的处理及提供赔偿的法律承担者和赔偿原则。在医学研究中,申办者应对于发生了与试验相关严重损害的受试者除积极救治外还须承担法律责任及相应的经济赔偿。待我国保险范围扩大及条件成熟后,申办者应对参加临床试验的受试者、研究者提供保险。以上风险处置内容应是伦理审查时的重要关注点,医学临床研究在保障受试者利益方面存在欠公正的倾向,[4]在伦理审查中往往发现,知情同意书包含的信息不够客观全面,有些知情同意书过多强调研究可能带来的受益,对可能出现的风险及处置一笔带过,用语含糊,尽量规避自身的责任;有的申办者递交的是保险公司对其公司的保险单,并没有申办者与受试者之间或申办者与医疗机构之间的保险承诺及知情同意书中对受试者风险处置的表述。因此,医学研究的伦理审查中要特别注意知情同意书中可能出现的疏漏,并要求知情同意书明确表明将受试者风险尽可能被控制在最小。
4 加强伦理委员会对医学研究知情同意的审查
为规避科学研究本身的风险以及科学研究成果的负面影响,科学研究必须通过伦理的审查考量,[5]对知情同意的审查是控制医学研究风险的重要环节之一,因此必须加大审查力度,维护受试者权益。尤其是在审查中要特别注意容易被忽视的要素如医学研究的背景简介、入选/排除标准、研究人员资历、研究过程、预期或非预期的不良反应、紧急情况时的救治方案、隐私保密以及重新获得知情同意的过程等。知情同意书撰写质量的提高,不仅需要伦理审查工作的加强,更需要制定完善统一的审查标准。知情同意审查标准不仅要涉及最基本的客观指标,如知情同意书的主要撰写要素,还要充分从受试人群考虑,涉及到主观的指标,如知情同意书语言是否容易理解,是否有歧义,是否含有暗示语言等。加强对伦理审查委员会委员的培训,促进其审查能力的提高,将有利于保护受试者权益,有利于伦理审查委员会的建设,有利于医学研究健康快速发展。
[1]国家食品药品监督管理局.药物临床试验质量管理规范(局令第3号)[Z].国家食品药品监督管理局,2003-8-6.
[2]国家食品药品监督管理局.关于印发药物临床试验伦理审查工作指导原则的通知[EB/OL].http://www.sfda.gov.cn/gsyjz10436/fj.rar,2010-11-02,2014-01-25.
[3]国家食品药品监督管理局.国家食品药品监督管理局器械司关于征求《医疗器械临床试验质量管理规范(征求意见稿)》意见的通知(食药监械函[2012]68 号)[EB/OL].http://www.sda.gov.cn/WS01/CL0027/74610.html,2012 - 08 -28,2014-01-25.
[4]胡晋红,黄瑾.医学研究伦理学的发展现状与前景[J].生命科学,2012,24(1):1250-1257.
[5]汪秀琴,熊宁宁,王思成.中医药临床研究与伦理审查[J].中国医学伦理学,2010,23(4):82 -83.
猜你喜欢
保健与生活(2022年16期)2022-08-06
科学与社会(2022年2期)2022-07-02
海外星云(2021年9期)2021-10-14
现代仪器与医疗(2021年1期)2021-06-09
大众健康(2020年7期)2020-08-25
中国医学伦理学(2019年11期)2019-01-17
小演奏家(2016年5期)2016-05-14
家庭用药(2016年4期)2016-04-23
分忧(2014年9期)2014-09-22
中国合理用药探索(2012年2期)2012-03-20
