
替米沙坦合成工艺研究进展
2022-10-22 10:26李维思曹亚运
广州化工 2022年18期
李维思,黄 双,曹亚运
(1 江苏中邦制药有限公司,江苏 南京 211300;2 南京红太阳医药研究院有限公司,江苏 南京 211300)
替米沙坦(Telmisartan),英文商品名为Micardis,化学名为:4′-[(1,4′-二甲基-2′-丙基[2,6′-二-1H-苯并咪唑]-1′-基)甲基]-[1,1′-二联苯基]-2-羧酸,CAS号:144701-48-4。替米沙坦是由德国Boehringer Ingelheim(勃林格殷格翰)公司开发,于1999年首次在美国上市,2000年在澳大利亚、比利时、英国上市,2002年在我国进口上市。替米沙坦是一种新型的降血压药物,是一种特异性血管紧张素Ⅱ受体(ATⅠ型)拮抗剂,用于治疗原发性高血压,既可以单独使用,也可以与其他抗高血压药物联合使用。2012年替米沙坦被批准用于年龄55岁及以上,存在发生严重心血管事件高风险且不能接受血管紧张素转换酶抑制剂(ACEI)治疗的患者,以降低其发生心肌梗死、卒中或心血管疾病导致死亡的风险。这是我国同类别血管紧张素Ⅱ受体拮抗剂(ARB)药物中首个被批准此项适应证的治疗药物。
替米沙坦制剂已在我国上市二十多年,原料药的生产也有十几年,经过不断优化改进,生产技术趋向成熟。近些年,围绕关键中间体的绿色化学技术开发、杂质研究等工作仍然非常活跃。
1 替米沙坦合成工艺
制备替米沙坦化合物专利为DE4137812,由德国Boehringer Ingetheim公司于1993年5月申请发明。该公司2004年又发表专利WO2004/087676,相应的中国专利为CN1768044A,对替米沙坦的制备方法进行了保护。此后,关于替米沙坦的制备工艺有多篇专利报道,据不完全统计,目前国内制备替米沙坦相关的工艺路线专利和文献约30多篇。目前国内合成替米沙坦的工艺路线概括如图1所示。
替米沙坦合成的工艺路线概括起来主要包括4类。路线一是以双咪唑和羧酸酯(如4′-溴甲基联苯-2-羧酸酯)为起始原料,经缩合、水解、酸化制备得到替米沙坦;路线二是以双咪唑和2-氰基-4′-溴甲基联苯为起始原料,经缩合,高温水解后酸化得到替米沙坦;路线三是以双咪唑和对溴溴苄经缩合反应后与邻硼酸钠或苯甲酸钠在醋酸钯催化下进行Suzuki偶联反应制备得到替米沙坦;路线四采用金属元素催化,后续加入二氧化碳制备替米沙坦。
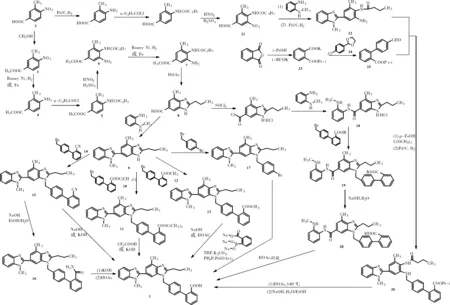
图1 替米沙坦合成路径图Fig.1 Synthesis route of Telmisartan
1.1 溴代联苯羧酸酯工艺
专利CN1344712是2001年由中国科学院上海药物研究所发表的专利,该专利报道的是采用双咪唑与4′-溴甲基联苯-2-羧酸酯(R=CH3、C2H5)经亲核取代反应得到替米沙坦的羧酸酯衍生物(R=CH3、C2H5),再水解脱保护得到替米沙坦[1]。同时文献(J.Med.Chem,1993,36(25):4040-4051)报道了双咪唑与4′-溴甲基联苯-2-叔丁酯反应,然后水解得到替米沙坦。上述方法合成路线较短,后处理简单,适合于工业化生产。
1.2 溴代氰基联苯工艺
WO2004/087676是由原研厂家德国Boehringer Ingetheim公司于2004年3月申请的一篇专利。该专利主要保护的是双咪唑与2-氰基-4′-卤代甲基联苯经缩合、高温水解制得替米沙坦[2]。和路线一相比较,该方法路线较为简单,采用的原材料2-氰基-4′-卤代甲基联苯相较于4′-溴甲基联苯-2-羧酸酯更易得,但氰基水解条件苛刻,需高温或加压进才能水解完全。
1.3 对溴溴卞偶联工艺
US20060264644报道了双咪唑与对溴溴苄经缩合,后以醋酸钯为催化剂,四氢呋喃/碳酸钾/三苯基膦共同作用下进行Suzuki偶联反应制备得到替米沙坦[3]。相较于上述两种路线,此路线较短,但是对溴溴苄和醋酸钯价格昂贵,制备成本比上述两种方法高,工业化生产有一定的局限性。
1.4 金属催化工艺
4-卤代甲苯与1,2-二卤代苯在金属元素例如镁、锂或锌的存在下反应,其中使用相对于1,2-二卤代苯0~0.9 mol,特别是0~0.2摩尔过量的4-卤代甲苯,通过卤素淬灭所产生的有机金属中间体。此外,也描述了如下方法:将所产生的2′-卤代-4-甲基联苯与2-(1-丙基)-4-甲基-6-(1′-甲基苯并咪唑-2-基)苯并咪唑偶联,得到3′-(2′-卤代-联苯-4-基甲基)-1,7′-二甲基-2′-丙基-1H,3′H-[2,5′]联苯并咪唑基,其可以进一步被转化为有机金属化合物,且将所述有机金属化合物与甲酸衍生物例如N,N-二甲基甲酰胺、烷基甲酸酯或二氧化碳进一步反应以获得替米沙坦[4]。
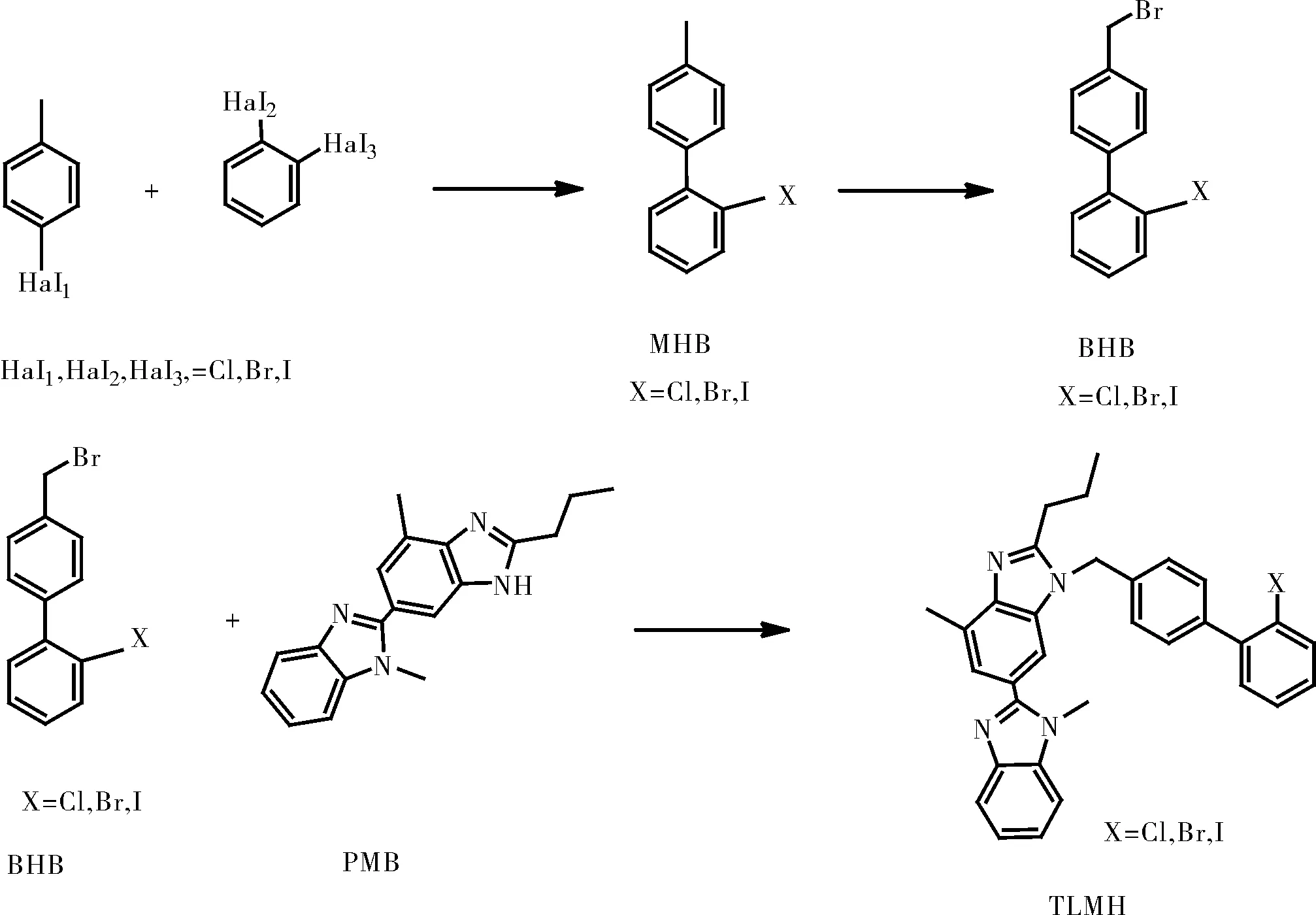
图2 通过对溴溴卞偶联工艺合成替米沙坦Fig.2 Synthesis of Telmisartan by p-Bromobromobian coupling process
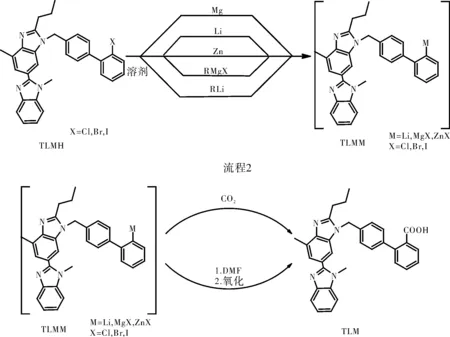
图3 通过金属催化工艺合成替米沙坦Fig.3 Synthesis of Telmisartan by metal catalytic process
2 替米沙坦关键中间体主链的合成工艺
关于替米沙坦的制备关键点是2-正丙基-4-甲基-6-(1′-甲基苯并咪唑-2-基)苯并咪唑即双咪唑的制备。为此,围绕双咪唑合成路线的相关专利和文献较多,方法也多种多样。按照起始物料的不同对关键中间体双咪唑的合成工艺总结如下:
2.1 以邻甲基苯胺为起始物料
以邻甲基苯胺为原料,经丁酰化、氯甲基化、Sommelet反应、环合、硝化、还原环合制得替米沙坦关键中间体双咪唑[5],再制备得到替米沙坦。
与上述工艺类似,以邻苯甲胺作为起始原料,与丁酰氯酰化得到(Z2),并通过氯甲基化生成(Z3),接着与六亚甲基四胺反应生成重要的中间体(Z4),而不是传统路线的需要通过皂化生成羧酸的甲酯化合物。化合物(Z4)经过硝化生成硝基化合物(Z5),用铁粉取代钯碳经过一步反应还原、环化得到化合物(Z7),再进一步和N甲基邻苯二胺反应得到2-正丙基-4-甲基-6-(1′-甲基苯并咪唑-2-基)苯并咪唑(双咪唑)[6]。
另一种方案也是以邻甲基苯胺为原料,先得到3-甲基-4-正丁酰胺基-5-硝基苯甲酸,与SOCl2反应成酰氯,与苯磺酰肼反应制备中间体1;中间体1被Zn,CaCl2和乙醇还原,再在醋酸中合环,制备中间体2;中间体2在碱存在下制成7-甲基-2-正丙基-3H-苯并咪唑-5-醛,即图4的化合物10[7]。
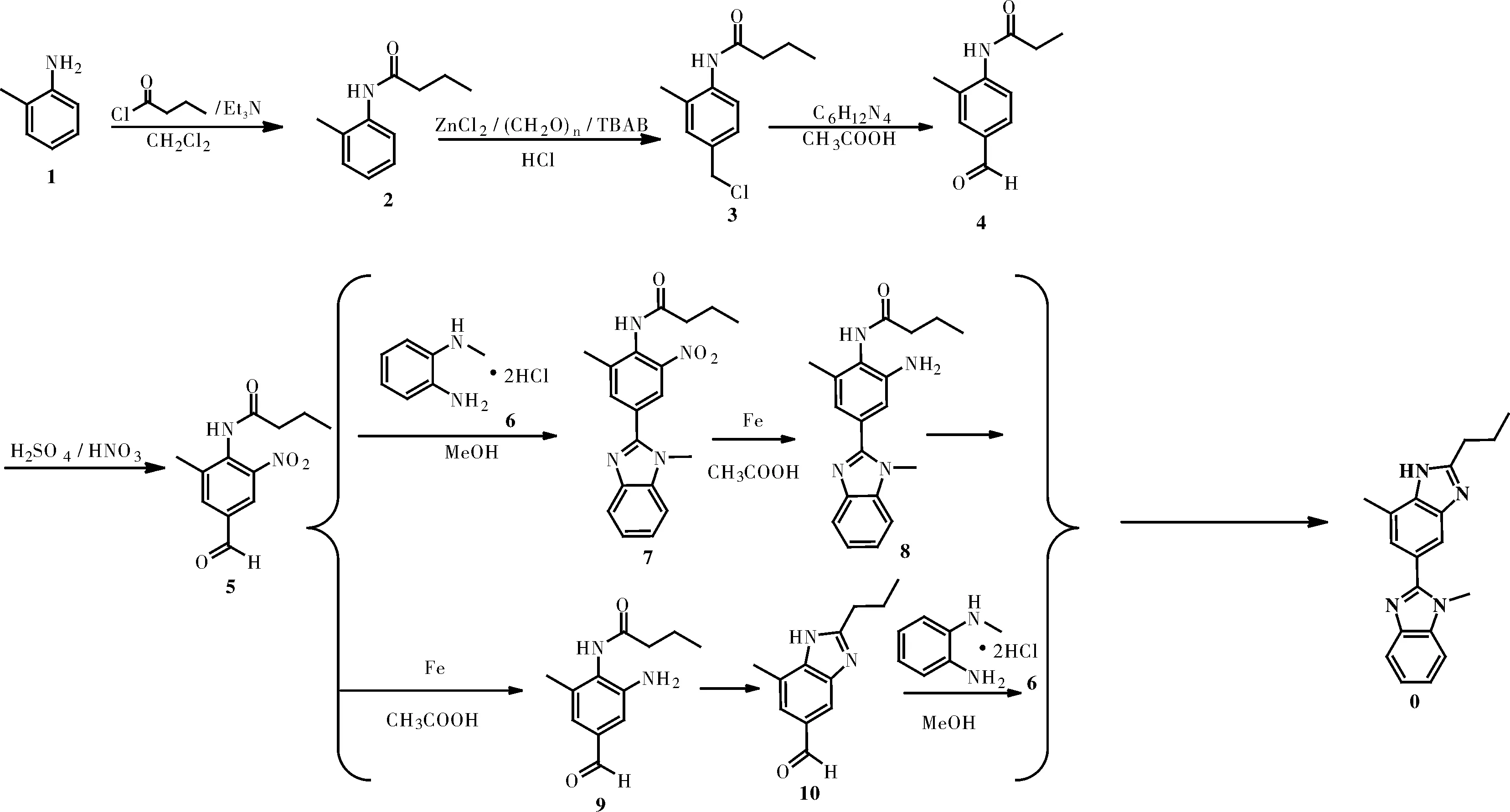
图4 以邻甲基苯胺为原料合成双咪唑Fig.4 Synthesis of Bisimidazole using o-Toluidine as raw material
在上述路线的基础上,为了避免上述工艺中存在的毒性大、腐蚀性强及致癌性的试剂和溶剂如氯化亚砜、硝酸、硫酸、多聚磷酸、二氯乙烷、乙二醇等,采用毒性小、安全好的试剂和溶剂如乙醇、甲醇、乌洛托品、空气、次氯酸钙、氢氧化钠等,更加绿色环保,对主链7的制备设计更符合绿色工艺的要求;对侧链18的合成工艺进行了优化,革除了毒性大、刺激性强的溴素以及昂贵的二溴海因和N-溴代琥珀酰亚胺,使用毒性小、安全好、价廉的氢溴酸作为溴代试剂,绿色环保,操作安全,工艺稳定[8]。
2.2 以3-甲基-4-氨基苯甲酸为起始物料
盐酸乙醇在0~35 ℃范围与丁腈、无水氯化氢作用,第一步所得在pH 5.0~11.0范围,与3甲基4氨基苯甲酸、冰醋酸混合,控温10~40 ℃反应,得式II中间体;式II中间体再与次氯酸钠溶液反应得2-正丙基-4-甲基-6-羧基苯并咪唑即单咪唑[9](此为合成双咪唑的前体)。
以3-甲基-4-氨基苯甲酸为起始原料,经酯化、酰化、硝化、还原、环合反应得到2-正丙基-4-甲基-6-羧基苯并咪唑[10];在多聚磷酸的作用下,与N-甲基邻苯二胺缩合,产物再与4’-溴甲基联苯-2-羧酸甲酯缩合,水解得到替米沙坦,总收率约为36%。
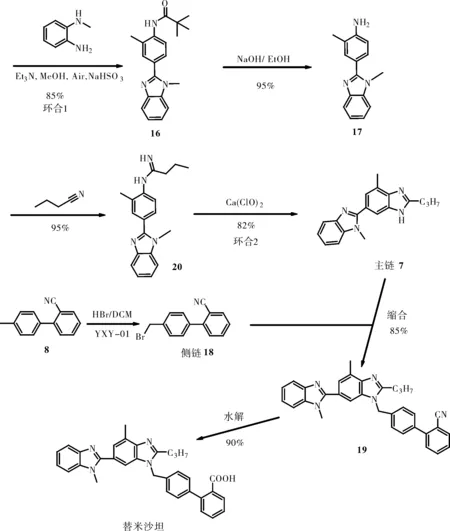
图5 以邻甲基苯胺为原料的改进方法合成双咪唑Fig.5 Optimization of synthesis of Bisimidazole using o-Toluidine as raw material
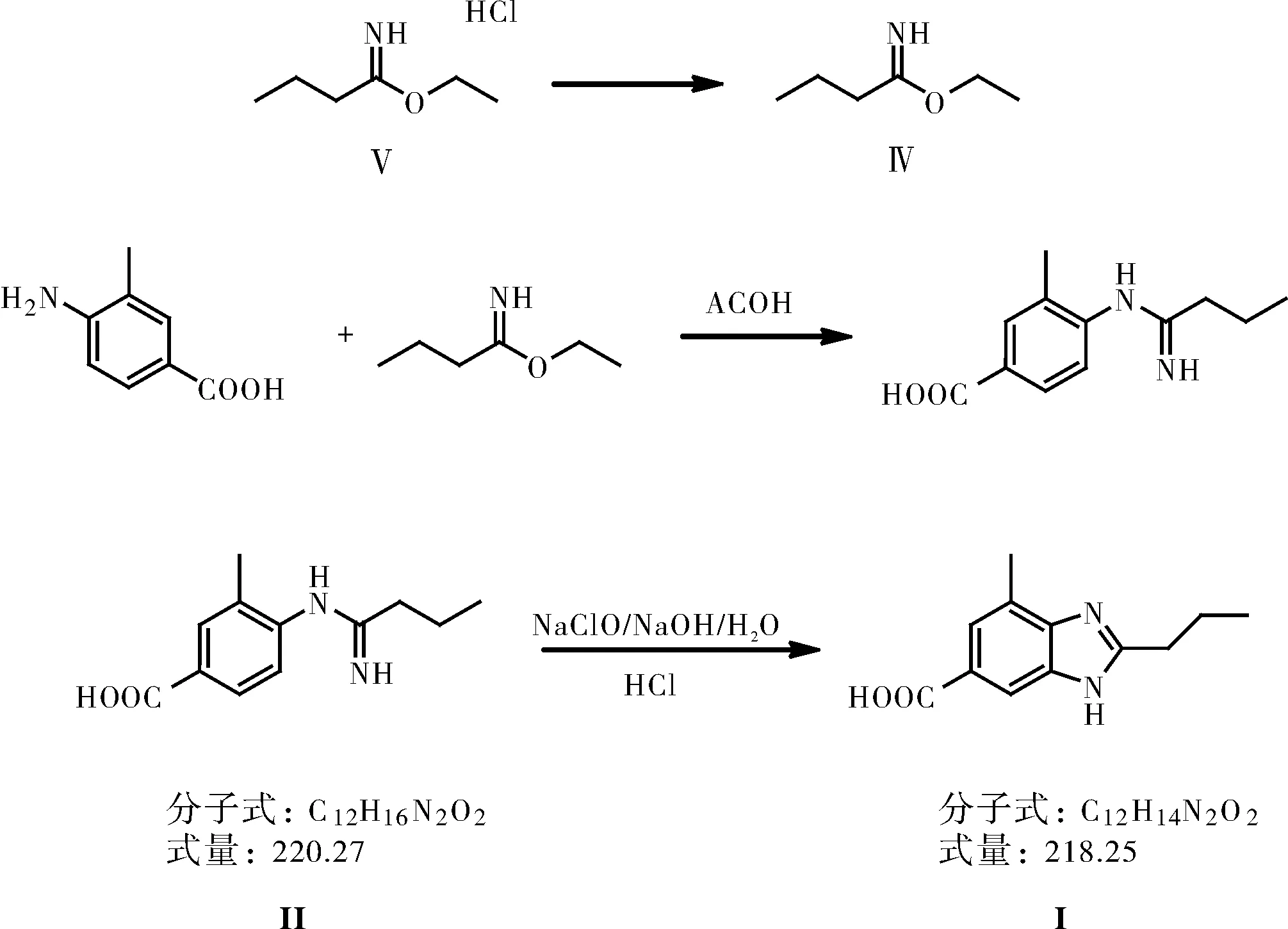
图6 以3-甲基-4-氨基苯甲酸为原料合成单咪唑Fig.6 Synthesis of monoimidazole using 3-methyl- 4-aminobenzoic acid as raw material
2.3 以3-甲基-4-硝基苯甲酸为起始物料
以3-甲基-4-硝基苯甲酸为起始原料,经过酯化、还原、酰化、硝化、还原、环合、水解,然后在多聚磷酸作用下与N-甲基邻苯二胺缩合反应,该方法为较为常见的双咪唑合成方法[11]。
2.4 以3-甲基-4-丁酰氨基-5-硝基苯甲酸甲酯为起始物料
以3-甲基-4-丁酰氨基-5-硝基苯甲酸甲酯为起始物料,醇类溶剂环境中,在硝基还原剂和水合肼共同参与下,在滴加温度为40~70 ℃、反应温度为40~70 ℃下进行还原反应制得,硝基还原反应的时间以检测反应完全为止,反应完全后,过滤旋干,得3-甲基-4-丁酰氨基-5-氨基苯甲酸甲酯[12],再进行后续环合等反应制备双咪唑。
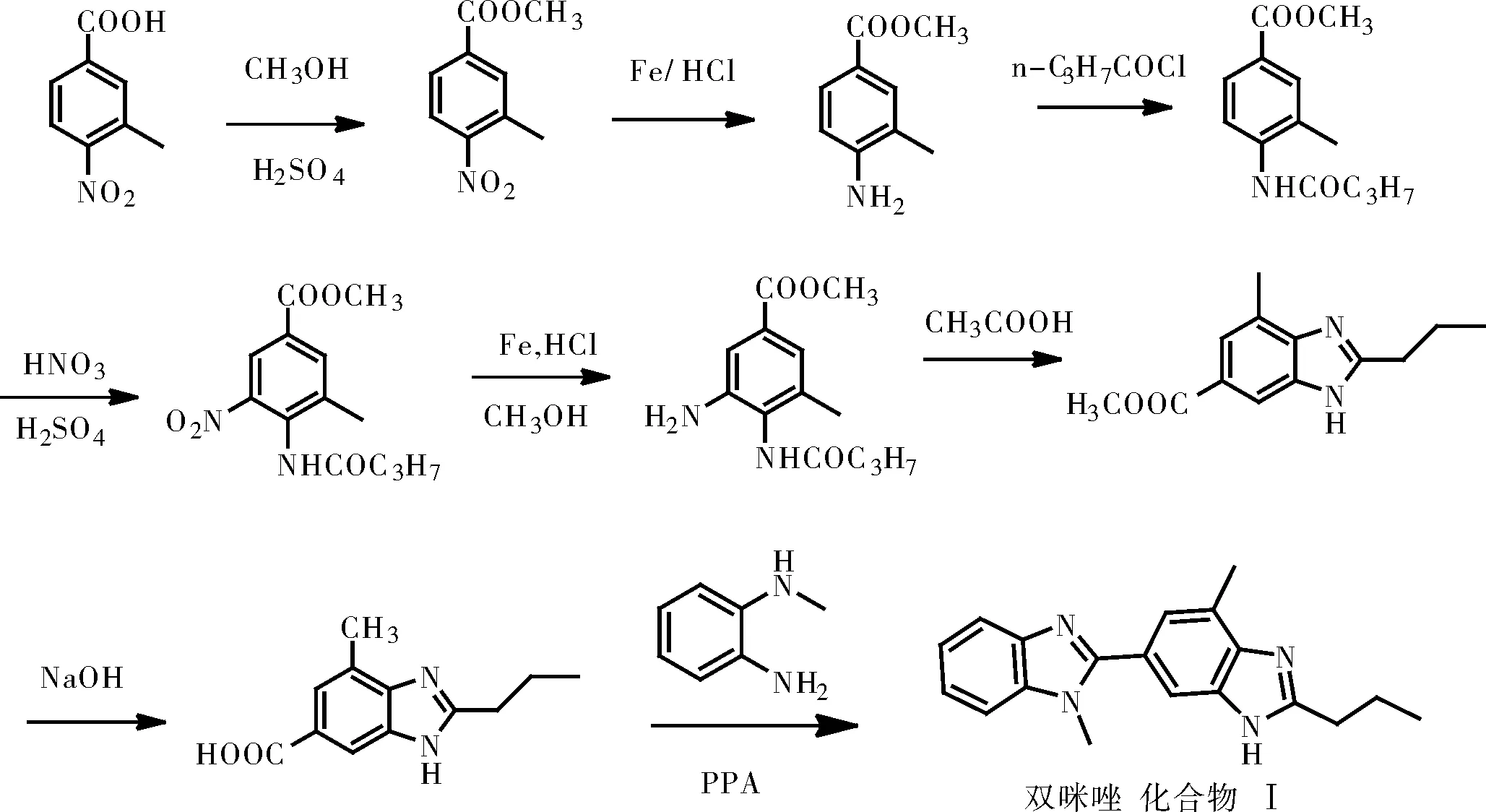
图7 以3-甲基-4-硝基苯甲酸为原料合成双咪唑Fig.7 Synthesis of Bisimidazole using 3-methyl-4-nitrobenzoic acid as raw material
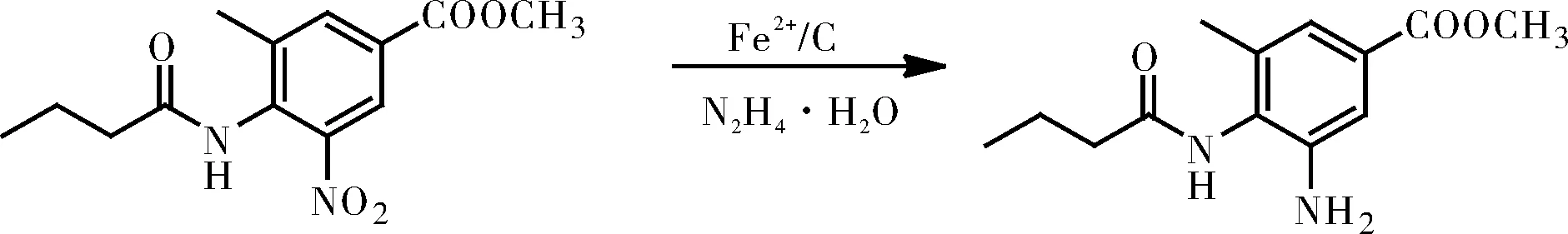
图8 以3-甲基-4-丁酰氨基-5-硝基苯甲酸 甲酯为原料合成双咪唑Fig.8 Synthesis of Bisimidazole using methyl 3-methyl-4-butyrylamino 5-nitrobenzoate as raw material
2.5 以N-(邻甲苯基)三甲基乙酰胺为起始物料
采用N-(邻甲苯基)三甲基乙酰胺和乌洛托品在三氟醋酸中通过Duff反应一步制备N-(4-甲酰基-2-甲基苯基)三甲基乙酰胺[13],再采用文献方法[14-16]制备得到替米沙坦。
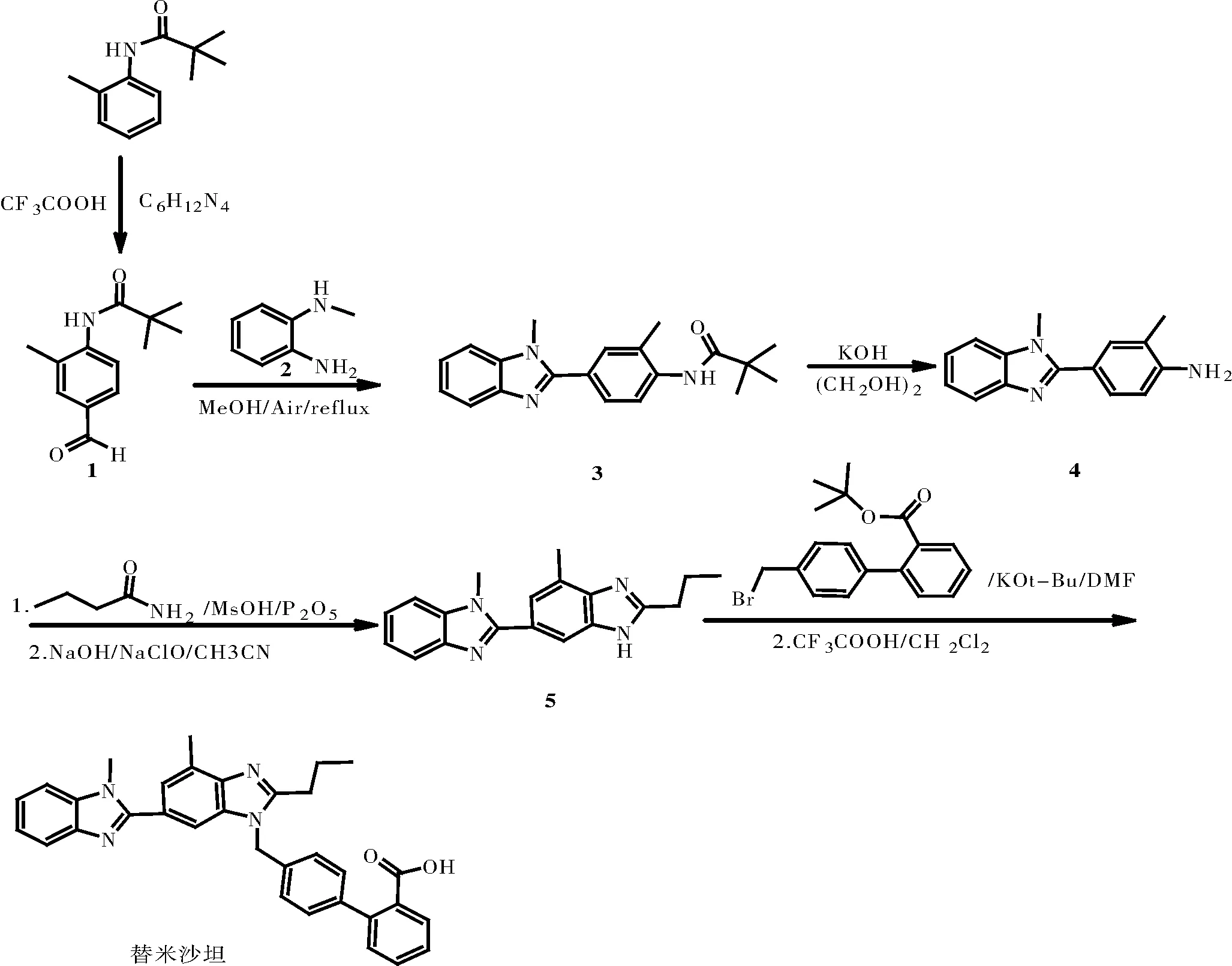
图9 以N-(邻甲苯基)三甲基乙酰胺为原料合成双咪唑Fig.9 Synthesis of Bisimidazole using N-(o-tolyl)trimethylacetamide as raw material
与上述反应类似,以N-(4-甲酰基-2-甲基苯基)三甲基乙酰胺和N-甲基1,2-苯二胺为原料进行反应,生成化合物14,再以化合物14为反应物,反应生成所述的替米沙坦中间体2-甲基4-(1-甲基-1H-苯并咪唑-2-基)胺(1)[17]。
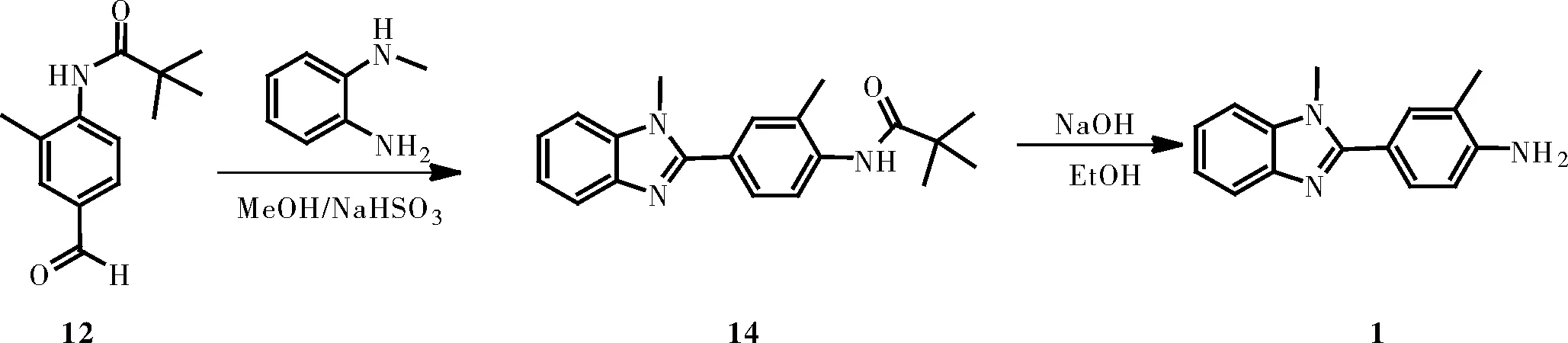
图10 以N-(邻甲苯基)三甲基乙酰胺为 原料的改进方法合成双咪唑Fig.10 Optimization of synthesis of Bisimidazole using N-(o-tolyl)trimethylacetamide as raw material
2.6 以2-甲基-6-硝基苯胺为起始物料
以2-甲基-6-硝基苯胺为原料,经NBS溴化、正丁酰氯在100 ℃下酰化、铁粉/氯化铵在乙醇和水中还原、醋酸介质中加热环合、Pd(dppf)Cl2催化100 ℃下插入羰基、氢氧化钠水解,共6步得到2-正丙基-4-甲基苯并咪唑-6-羧酸,总收率达62%[18]。
3 替米沙坦中间体侧链的合成工艺
以对氯甲苯和邻氯苯腈为原料,对氯甲苯和镁条先制成格氏试剂,然后在无水二氯化锰的催化下,通过格氏反应,和邻氯苯腈进行偶联反应,制得替米沙坦重要的侧链中间体4′-甲基-2-氰基联苯[19]。对合成条件进行了优化和改进,以邻氯苯腈计,目标产物的收率为70%,高于文献报道的56%。以3-甲基-4-氨基苯甲酸为起始原料,与甲醇在80 ℃下反应,生成3-甲基-4-氨基苯甲酸甲酯,产率92%;以氯苯为溶剂,3-甲基-4-氨基苯甲酸甲酯与正丁酰氯在75 ℃下反应合成3-甲基-4-丁酰氨基苯甲酸甲酯,产率95%;3-甲基-4-丁酰氨基苯甲酸甲酯与发烟硝酸与浓硫酸(3∶2)组成的混酸在-5 ℃下反应,合成3-甲基-4-丁酰氨基-5-硝基苯甲酸甲酯;3-甲基-4-丁酰氨基-5-硝基苯甲酸甲酯以钯碳作为催化剂常压加氢还原生成3-甲基-4-丁酰氨基-5-氨基苯甲酸甲酯,产率93%;3-甲基-4-丁酰氨基-5-氨基苯甲酸甲酯在盐酸的作用下关环生成4-甲基-2-正丙基-6-甲氧羰基苯并咪唑,最后在氢氧化钠的作用下水解得4-甲基-2-正丙基-6-羧基苯并咪唑。六步总收率54.0%。
4 替米沙坦结构类似物的合成工艺
除了工艺研究,在原料药合成过程以及药品研发注册中,杂质谱的研究和杂质控制都非常重要。采用4-氯-2-硝基-N-甲基苯胺为原料,经过还原、环合、缩合及水解酸化等过程,可以得到替米沙坦结构类似物,即替米沙坦原料药的关键杂质J[20]。
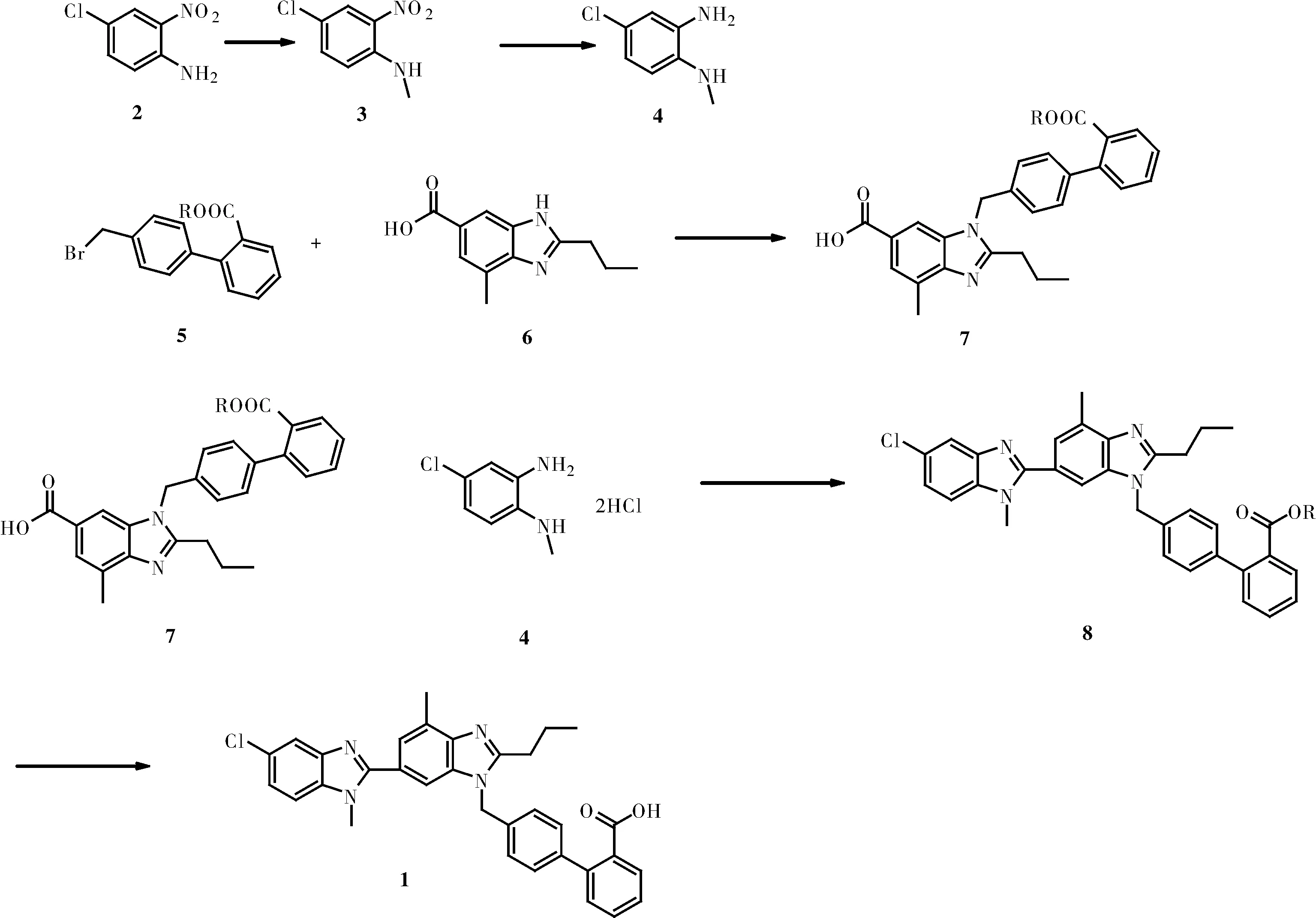
图11 替米沙坦结构类似物的合成Fig.11 Synthesis of Telmisartan structural analogs
5 结 语
总结了替米沙坦的四种合成工艺,描述了替米沙坦关键中间体主链双咪唑的的六种合成路线和相应工艺情况,概述了替米沙坦侧链氰基联苯以及替米沙坦关键杂质J的合成工艺。随着安全环保要求的不断提升,上游原材料价格的上涨,下游制剂带量采购的限价,替米沙坦原料药和关键中间体的工艺研究和改进仍有很多工作值得深入开展。
猜你喜欢
中国药学药品知识仓库(2022年10期)2022-05-29
小作家报·教研博览(2022年11期)2022-04-02
食品界(2019年4期)2019-05-05
山东工业技术(2018年9期)2018-05-26
百科知识(2016年22期)2016-12-24
科技视界(2016年26期)2016-12-17
百科知识(2016年16期)2016-10-29
家庭百事通·健康一点通(2016年7期)2016-08-04
股市动态分析(2015年12期)2015-09-10
中小企业管理与科技·中旬刊(2014年10期)2015-02-03
