
Ⅰ型戈谢病影像表现一例
2022-06-30 14:26陈方方齐先龙
影像诊断与介入放射学 2022年3期
陈方方 齐先龙
戈谢病(Gaucher’s dieasease,GD)是一种罕见的常 染色体隐性遗传病,发病率约为十万分之一至五万分之一[1],是一类由葡萄糖脑苷酯酶(glucocerebrosidase,GBA)缺乏引起的溶酶体蓄积病。可累及骨骼、肝脏、脾脏和中枢神经系统。目前以骨骼、肝大、脾大受累为主要表现的病例报道居多,以脾大并脾内多发结节灶的病例报道较少见,本文分享一例以脾大并发脾内多发结节灶为主要表现的戈谢病患者的影像表现。
病例资料患者,男,44 岁,体检时发现脾大,血小板减少,偶有乏力、腹胀,7 d 前无明显诱因出现头晕,门诊以脾大,血小板减少收入院。入院后血常规示:血小板97×10-9/L,网织红细胞计数129.4×10-9/L,血红蛋白138 g/L。复查血常规示:红细胞3.66×10-12/L,白细胞2.88×10-9/L,中性粒细胞1.5×10-9/L,血红蛋白103 g/L,血小板51×10-9/L。腹部CT 示(图1):脾明显增大,平扫密度轻度不均匀性减低,增强扫描脾内可见多发大小不等结节状低密度灶,边缘不清,呈轻度不均匀性延迟强化。MPR 重组冠状位脾内结节灶显示更清晰。肋骨呈磨玻璃密度改变。骨盆、双侧股骨骨质不均匀性减低,股骨骨髓腔膨大。CT 诊断:考虑脾淋巴瘤。腹部MRI 示(图2):脾脏明显肿大,其内见弥漫分布的大小不等结节样、团块状异常信号灶,T1WI 呈低信号、T2WI呈低信号,其内出现类似“轮辐征”的表现;正相位结节灶可见信号较低,反相位未见明显减低;DWI(b=800 s/mm2)信号减低,ADC 未见明显增高。双侧髋关节X 线平片示双侧髋关节组成骨及双侧股骨干骨质密度不均匀性减低,呈网格状改变,骨质稀疏,骨小梁破坏,骨髓腔膨胀扩大,骨皮质变薄(图3)。患者完善溶酶体贮积基因检测:GBA基因突变阳性:c.907C>A(p.Leu303Ile)杂合,GBA 活性:4.23 nmoL/(h·mgPro),较正常[7.55~10.19 nmoL/(h·mgPro)]明显减低,诊断为戈谢病,患者无该病家族史。
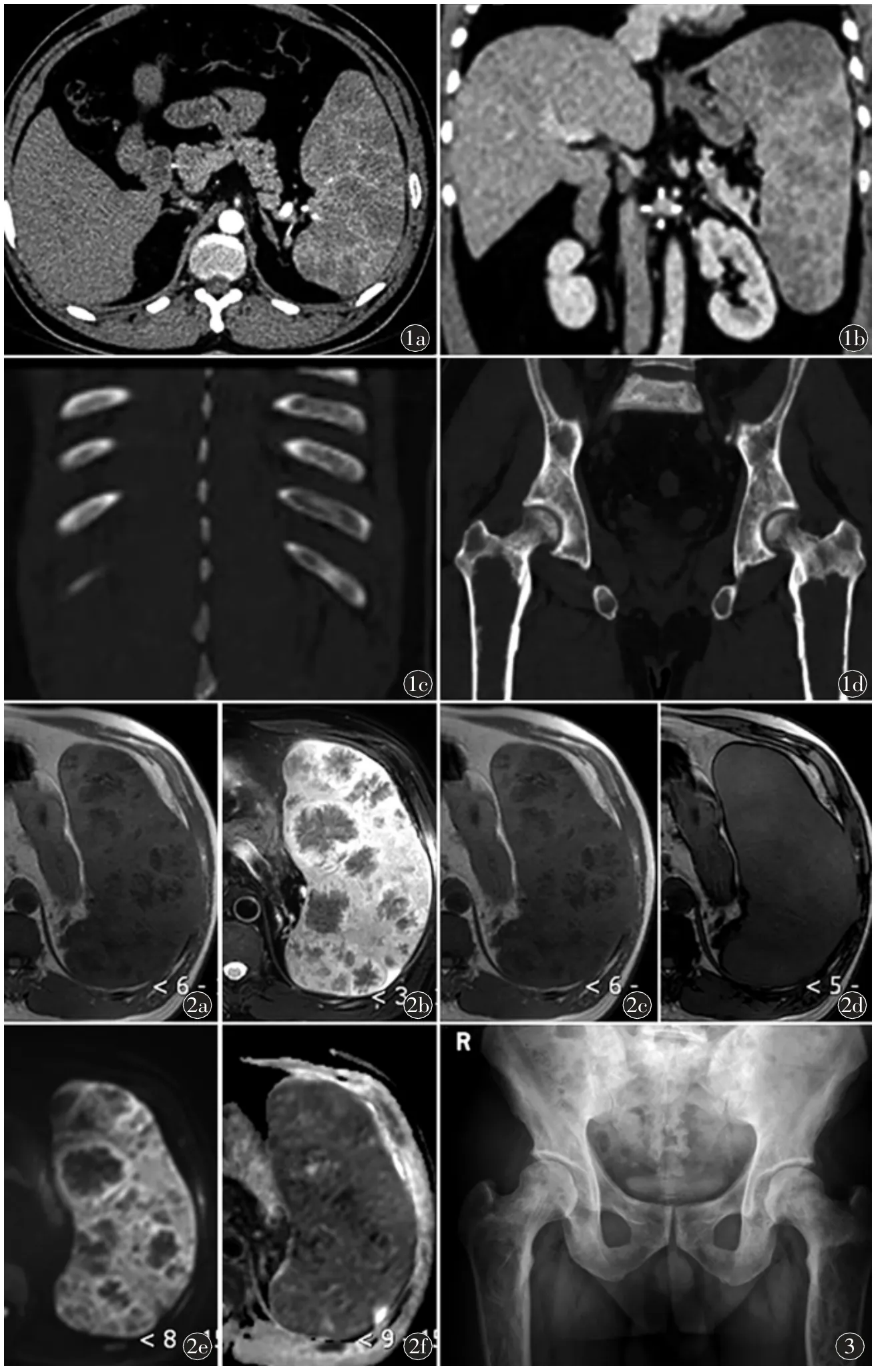
图1 a)CT 增强动脉期轴位示脾脏体积增大,脾内见多发大小不等低密度结节状灶,边缘不清;b)MPR 重组冠状位对脾内结节灶显示更清晰;c)肋骨呈磨玻璃密度改变;d)MPR重组冠状位示骨盆及双侧股骨骨质不均匀性减低,骨髓腔膨大图2 腹部MRI 示脾内见弥漫性分布的大小不等结节样、团块状异常信号灶。a)T1WI 呈低信号;b)T2WI 呈低信号,其内出现类似“轮辐征”的表现;c)正相位示结节灶可见信号减低;d)反相位未见明显减低;e)DWI(b=800 s/mm2)信号减低;f)ADC 未见明显增高图3 双侧髋关节X 线片示双侧髋关节组成骨及双侧股骨干骨质密度不均匀减低,骨质稀疏,骨小梁破坏,骨髓腔膨胀扩大,骨皮质变薄
讨论戈谢病是由于GBA 基因缺陷,导致其底物葡萄糖脑苷脂在巨噬细胞溶酶体中蓄积,这些病理性的巨噬细胞被称为戈谢细胞,戈谢细胞浸润肝、脾、骨骼及肺,甚至神经系统,继而引起肝脾肿大、血细胞减少、骨骼异常及神经系统症状[2]。根据临床表现可分为三种类型。Ⅰ型(非神经病变型):此型最常见,占90%以上,主要表现为肝脾肿大、脾功能亢进、血小板减少和贫血、骨痛等。其次还会出现生长发育迟缓,肺动脉高压[3]及胆囊炎、胆石症的表现。Ⅱ型(急性神经病变型):婴幼儿时期发病,主要表现为神经系统受累,如癫痫发作、斜视、喉痉挛、吞咽异常等,可伴有Ⅰ型的症状,一般2~4 岁死亡[4]。Ⅲ型(亚急性神经病变型):该型临床症状变化较大,儿童期发病,除了有肝脾肿大、骨受累外,还会出现神经系统受累表现,伴发育迟缓,学习障碍、痴呆等,病程进展缓慢[5]。Ⅲ型又分为三种亚型:Ⅲa 型以快速进展的神经系统症状为主要表现;Ⅲb型主要表现为骨骼受累,中枢神经系统症状较少;Ⅲc 型主要表现为心脏瓣膜钙化。欧美和中东国家的患者中,Ⅰ型占90%以上。本例患者主要表现为脾大,血小板减少,贫血,骨骼病变,无神经系统异常表现,符合Ⅰ型戈谢病。
戈谢病诊断金标准为:患者外周血白细胞或皮肤成纤维细胞中GBA 活性降低至正常值的30%以下时,可确诊[6,7]。对于以骨骼改变为主要表现的戈谢病患者,常会进行骨髓穿刺检查,但需要注意的是骨髓形态学检查发现“戈谢细胞”,并不能完全确诊戈谢病,因为其他疾病也会出现类似细胞,如其他类型的溶酶体蓄积病或慢粒白血病[7]。本例患者GBA 活性低于正常值的44%,且GBA 基因突变阳性:c.907C>A(p.Leu303Ile)杂合,即GBA 基因中的第907位的胞嘧啶被腺嘌呤取代,使第303 位的亮氨酸变为异亮氨酸,因此得以确诊为戈谢病。
本例患者较典型的表现是脾内多发结节灶,此前曾有文献报道[8]一例脾内多发结节灶案例,其术后病理证实结节灶为大量载脂巨细胞(戈谢细胞)大量聚集,呈“巢团状”分布。也有诸多文献报道戈谢病患者术后脾脏切片铁染色(+),血中铁蛋白含量高。本例未行骨髓穿刺及脾切除术,但脾内结节灶在磁共振上表现为T1WI 低信号、T2WI 低信号,正相位信号减低,反相位未见明显减低,符合铁沉积的磁共振表现;结节灶内具有类似“轮辐征”的影像表现。因此结合文献报道,推测脾中低密度结节灶为含铁的戈谢细胞不同程度堆积所致。
多数戈谢病患者骨骼受侵,可累及任何骨,渐进且不可逆。戈谢病的骨受累包括骨痛、骨危象、缺血性坏死、骨折,最新共识[8]认为骨痛和脊柱后凸是主要症状。X 线摄影表现为骨质疏松,骨皮质变薄,髓腔扩大,也可发生骨质硬化。长骨干骺端烧瓶样畸形是特征性的表现。磁共振征象:早期骨髓T1WI、T2WI 信号减低,压脂T2WI 信号增高,骨梗死时可表现为混杂信号。本例患者股骨远端未见明显烧瓶样改变,而是以网格状骨质密度不均匀性减低为主要特征。
本病目前治疗方法包括酶替代疗法、底物减少疗法、骨髓移植、脾切除等。首选疗法是酶替代疗法,目前市场用药为伊米苷酶、维拉苷酶α。主要用于Ⅰ型和Ⅲ型戈谢病的治疗。有研究指出酶替代疗法对内脏损害有可逆作用,它可能减少肝脾肿大,最小化骨骼病变,纠正贫血和血小板减少,并优化生长[9]。2021 年中国儿童戈谢病诊治专家共识[7]指出:戈谢病一般不建议行脾切除术,虽然切除脾脏可解除脾亢,但是可能会加重肝脏、肺、骨骼系统症状。本例患者未行脾切除治疗,而选择服用伊米苷酶治疗。
回顾本例的影像表现,需要与脾内淋巴瘤及转移瘤相鉴别,脾内淋巴瘤增强扫描可呈“地图样”不均匀强化,但在磁共振上呈T1WI 稍低信号、T2WI 稍高信号,特别是DWI 呈明显高信号,ADC 值减低,所以不考虑为脾内淋巴瘤。脾内转移瘤影像表现不一,磁共振上常表现为T1WI 低信号、T2WI 高信号,周围可出现水肿信号,脾内转移瘤常可并发肝脏或其他部位的转移,且本例患者行影像学检查,未发现原发肿瘤病灶,所以也不考虑为转移瘤。综合本例患者临床及影像资料,提示遇到血常规三系(红细胞、白细胞、血小板)减少或仅表现为血小板减少合并肝脾肿大,脾内结节灶,骨质密度不均匀性减低患者,应联想到本病,询问患者家族史,联合临床尽早行GBA 活性检测及相关基因检测。
综上所述,本病是较罕见的常染色体隐性遗传病,对于有戈谢病家族史者应注意完善产前基因筛查,对于有相关临床症状患者应联想到本病,及时行影像学、生化学、遗传学检查,及早酶替代治疗,改善预后。
猜你喜欢
妇女生活(2022年5期)2022-06-09
体育科技文献通报(2022年3期)2022-05-23
初中生学习指导·提升版(2020年5期)2020-09-10
青年文学家(2020年16期)2020-07-13
文苑(2018年18期)2018-11-08
中外医疗(2016年33期)2017-03-02
诗林(2016年5期)2016-10-25
数理化学习·高三版(2015年3期)2015-10-21
中国医药科学(2014年24期)2015-03-16
职业·中旬(2009年12期)2009-06-01
