
一例骨骼发育异常胎儿ALPL 基因的遗传学分析
2022-02-27 08:12唐克峰夏莉花沈国松
浙江中西医结合杂志 2022年2期
唐克峰 李 志 夏莉花 舒 艳 胡 刚 方 嵘 沈国松
低磷酸酯酶症(hypophosphatasia,HPP)是一种由于编码组织非特异性碱性磷酸酶(tissue non-specific alkaline phosphatase,TNSALP)的碱性磷酸酯酶基因(alkaline phosphatase gene,ALPL)变异导致的罕见常染色体显性或隐性遗传病,临床表现主要是骨骼和牙齿矿化不全以及血清碱性磷酸酶(ALP)活性偏低[1]。重症HPP 的发病率大概为十万分之一,轻度HPP 较多见,血清ALP 活性和分子遗传学是最常用的诊断方法,目前尚无可靠的治疗方法[2]。本研究中,我们对1 例骨骼发育异常胎儿的染色体核型、全基因组拷贝数变异以及骨骼系统相关基因的致病性变异进行分析,为其产前诊断提供分子遗传学依据。
1 资料与方法
1.1 一般资料 孕妇,24 岁,停经19+2W,唐氏筛查高风险(1/180),产前诊断超声(超声仪器是GE,volusionE8,超声频率3.5~5MHz,检查方法:超声探头寻找胎儿上肢,显示完整肱骨,尽量让声束与肱骨垂直,正常肱骨是平直的,成角的肱骨显示肱骨分成两条有角度的强回声。股骨也是同样的方法显示。)显示股骨及肱骨弯曲成角(见图1)。余无殊。夫妻否认近亲结婚,家族中无类似病史。所有研究对象均已签署知情同意书。本研究经本院伦理委员会批准(2019-R-016)。
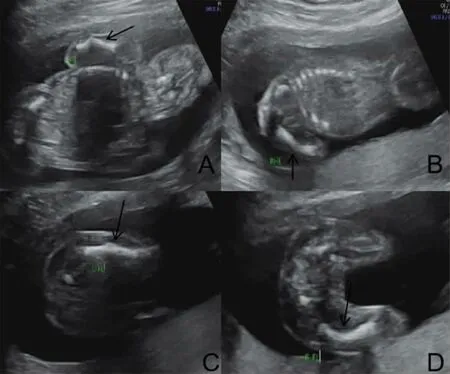
图1 胎儿产前诊断超声图(A:胎儿左肱骨成角;B:胎儿右肱骨成角;C:胎儿左股骨成角;D:胎儿右股骨成角)
1.2 方 法
1.2.1 常规染色体核型分析 无菌条件超声引导下经腹壁行羊膜腔穿刺术,抽取羊水细胞30mL,并对羊水细胞进行培养、制片、G 显带核型分析。
1.2.2 染色体微阵列分析技术(chromosomal microarray analysis,CMA)检测 利用Affymetrix Cytoscan 750k 芯片(CYTOSCAN 750K ARRAR AND R p/n 901859)检测羊水细胞全基因组不平衡现象。
1.2.3 应用DNA 提取试剂盒(厦门至善生物公司,批号200517)提取胎儿、孕妇及其丈夫外周血的基因组DNA,利用紫外分光光度计测量待检DNA 浓度及纯度,取一定量的DNA 原液稀释成20ng/μL 的工作浓度,-20℃保存备用,DNA 原液-70℃保存。使用Aligent SureSelect 方法制备检测样本。文库采用Illumina HiSeq X-Ten 系统完成高通量测序。利用FastQC、BWA(v0.7.15-r1140)、Picard、GATK(v3.7-0)、Annovar 及VEP 等软件包进行生物信息学分析。结合人群基因变异数据库(dbSNP 数据库(SNP150)、千人基因组数据库等)信息,去除MAF>0.01 或MAF>0.05 的高频变异。参考美国医学遗传学与基因组学学会(American College of Medical Genetics and Genomics,ACMG)相关指南[3],并筛选出与先证者表型相关的可疑致病性变异。针对筛选出的可疑致病基因变异,利用Primer 3 软件设计PCR 引物进行扩增及Sanger 测序分析。采用Mutation Surveyor等软件对测序数据进行分析。孕妇及其丈夫、胎儿DNA 样本测序结果进行家系共分离分析。
2 结果
2.1 常规染色体检型分析结果 胎儿羊水染色体核型分析结果为46,XN。
2.2 CMA 检测结果 未发现致病性拷贝数变异、杂合性缺失及单亲二倍体。
2.3 基因检测结果 提示胎儿ALPL 基因存在第2外显子c.18del 和第9 外显子c.979T>C 复合杂合变异(见图2)。其中c.18del 为移码变异,属于功能缺失性变异,功能预测软件结果偏向于致病性变异,根据ACMG 指南[3]分析该变异致病性为P(致病性变异)=PM2(ESP 数据库、千人数据库、EXAC 数据库中正常对照人群中未发现的变异)+PM3(在隐性遗传病中,在反式位置上检测到致病变异)+PVS1(当一个疾病的致病机制为功能丧失时,无功能变异)。c.979T>C为错义变异,功能预测软件结果偏向于致病性变异,预测值为0.991。根据ACMG 指南[3]分析该变异致病性为P(致病性变异)=PM2(ESP 数据库、千人数据库、EXAC 数据库中正常对照人群中未发现的变异)+PM3(在隐性遗传病中,在反式位置上检测到致病变异)+PS3(体内、体外功能实验已明确会导致基因功能受损的变异)+PP3(多种统计方法预测出该变异会对基因或基因产物造成有害的影响,包括保守性预测、进化预测、剪接位点影响等)。ALPL 基因转录本为NM_000478.4。

图2 碱性磷酸酯酶基因Sanger 测序示意图
2.4 孕妇夫妇相关基因验证结果 孕妇为c.18del杂合变异携带者,其丈夫为c.979T>C 杂合变异携带者(见图2)。
3 讨论
HPP 是一种罕见遗传性代谢性骨病,不同的基因变异位点对于酶的生物学功能影响程度不一导致临床表现多种多样[4]。根据患者最早出现症状的年龄和严重程度将HPP 分为围产期致死型、围产期良性型、婴幼儿型、儿童型、成人型和牙齿型,鉴于目前没有根治方法,该病的遗传咨询和产前诊断显得尤为重要。其中围产期致死型表现出严重的骨骼矿化障碍,胎儿存在发育迟缓、骨骼成角等骨骼畸形,通常导致死胎或新生儿死亡[5]。
ALPL 是HPP 的致病基因,位于染色体1p36.1,包含12 个外显子,编码524 个氨基酸,在基因组中跨度约50kb,其mRNA 长度为2580bp,开放读框ORF 长度为1575bp。截至2020 年5 月29 日,国际上建立的低磷酸酯酶症基因变异数据库(http://www.sesep.uvsq.fr/03_hypo_mutations.php)已报道了410 个ALPL 基因变异,变异类型绝大部分为错义变异共292 种(占71.2%),缺失变异有45 种(占11.0%),其它还包括剪切变异、插入变异、无义变异等多种变异类型,变异分布于ALPL 基因各编码区,在外显子5区域发生变异频率最高。Mornet[6]研究发现HPP 患者ALPL 基因变异中9 号外显子变异相对较多,提示可能存在种族差异。95%的HPP 可检测到ALPL 基因变异,即使未检测到ALPL 基因变异,也有可能变异位于内含子区或调节区,当然也可能存在新的致病基因[7]。ALPL 基因编码的TNSALP 是一种广泛存在于各类组织的磷酸单酯酶,在骨骼矿化作用中表现为催化和水解焦磷酸并产生羟磷灰石结晶需要的磷,该酶活性降低可以引起无机焦磷酸酶盐等多种底物蓄积,从而引起骨软化或者佝偻病[8]。
本研究中对1 例胎儿超声发现骨骼成角异常的孕妇进行侵入性产前诊断,其中羊水细胞染色体核型为46,XN,羊水细胞CMA 检测未发现致病性拷贝数变异、杂合性缺失及单亲二倍体。骨骼系统相关基因检测提示胎儿ALPL 基因存在第2 外显子c.18del和第9 外显子c.979T>C 复合杂合变异。其中c.18del为移码变异,属于功能缺失性变异,该变异导致ALPL 基因编码的第7 位氨基酸缬氨酸变成酪氨酸,且继续编码12 个氨基酸后终止翻译,产生的截断蛋白丧失了发挥酶活性及骨骼矿化作用至关重要的区域。该变异十分罕见,仅有刘海娟等[9]报道了1 例儿童型低磷酸酶血症患者存在c.18del 和c.G407C 复合杂合变异,但c.G407C 变异临床意义不明确,患儿临床表现为门齿脱落,双侧桡骨、右股骨弯曲畸形,右下肢较左下肢短等,血ALP 19U/L(正常值42~390U/L),长骨弯曲表现与本研究一致。另一个变异c.979T>C 为错义变异,功能预测软件结果偏向于致病性变异,导致ALPL 基因编码的第372 位氨基酸苯丙氨酸变成亮氨酸,从而影响了酶的活性。该变异有多例HPP 患者文献报道,其中有13 例是c.979T>C与c.1559delT 致病性变异形成的复合杂合变异[10],并且功能实验提示该变异会导致酶的活性降低至野生型的70%左右[11]。研究表明,在HPP 患者中观察到的表型异质性极高,主要是由于ALPL 基因的错义变异导致残余酶活性不同引起的[12]。上述ALPL 基因两个变异分别来自父母双方,符合常染色体隐性遗传病,结合胎儿超声提示的股骨和肱骨各自成角表现,诊断胎儿为首例c.18del 和c.979T>C 复合杂合变异的HPP。由于严重的围生期致死型,可以引起骨骼矿化异常而导致死产,或者出生后新生儿期因骨骼畸形、钙磷代谢异常、广泛矿化不良或者肺发育不良而死亡[13]。孕妇最终选择引产,引产后我们对死胎进行全身X 线检查,显示肱骨、尺骨、桡骨以及股骨、胫骨皆有成角弯曲现象(见图3),提示预后不良。而产前诊断超声只显示肱骨和股骨弯曲,可能是由于胎儿在体内体位原因导致某些骨骼异常显示不清。由于该病呈常染色体隐性遗传方式,所以本例夫妇再生育胎儿患病风险为25%,可尝试自然怀孕后适时产前诊断或者直接选择第三代试管获得健康婴儿。
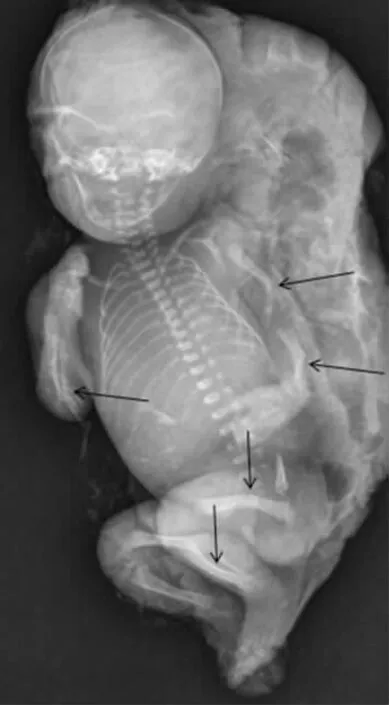
图3 死胎全身X 线图(四肢长骨弯曲成角)
综上所述,ALPL 基因存c.18del 和c.979T>C 复合杂合变异很可能是这例HPP 胎儿的骨骼异常原因,为HPP 的遗传咨询以及产前诊断提供了可靠的依据。
猜你喜欢
安徽农业大学学报(2022年2期)2022-11-09
中华骨与关节外科杂志(2022年1期)2022-08-31
临床骨科杂志(2022年3期)2022-06-23
昆明医科大学学报(2022年2期)2022-03-29
考试周刊(2017年26期)2017-12-12
校园英语·下旬(2017年7期)2017-07-14
科技视界(2016年27期)2017-03-14
中学生理科应试(2016年7期)2016-05-14
江苏农业科学(2015年11期)2016-01-27
中国动物保健(2015年4期)2015-10-21
