
HPV16 E7通过调控miR-29a-CDK6信号通路促进肿瘤细胞增殖*
2022-01-16 10:34熊焱强刘朝奇李志英
华中科技大学学报(医学版) 2021年6期
熊焱强, 冯 晶, 刘朝奇, 李志英
1三峡大学附属仁和医院妇产科,宜昌 443001 2三峡大学肿瘤微环境与免疫治疗湖北省重点实验室,宜昌 443002
宫颈癌(cervical cancer)是最常见的妇科恶性肿瘤之一,2018年全球约有57万新发病例,31.1万死亡病例,在女性恶性肿瘤中发病率排第2位,死亡率排第4位[1]。高危型人乳头瘤病毒(human papillomavirus,HPV)持续感染是宫颈癌的主要发病原因,其中以HPV16、18型与宫颈癌的相关性最强,而E6和E7是HPV的主要癌基因[2]。研究发现,HPV可以通过早期表达的调节蛋白作用于宿主细胞的miRNA引起宿主细胞增殖、凋亡、侵袭等生物学行为改变[3]。在前期研究中我们发现HPV16 E7可以促进肿瘤细胞增殖,加速细胞周期G1期向S期进展,其机制可能与下调miR-29a的表达有关[4]。本文旨在探究HPV16 E7基因通过调控细胞内miR-29a表达促进细胞增殖的分子机制,为进一步研究宫颈癌的发病机制及新的治疗靶点提供初步理论依据。
1 材料与方法
1.1 主要试剂及仪器
主要试剂:RPMI 1640培养液(美国Sigma公司),小牛血清(美国Gibco公司),Trizol(美国Invitrogen公司),反转录试剂盒(美国Fermentas公司),PVDF膜、蛋白印迹相关试剂(美国Amersham公司),鼠抗人CDK6抗体、鼠抗人β-actin抗体(美国Santa Cruz公司),羊抗鼠IgG-HRP抗体(北京中杉金桥生物技术公司),双荧光素酶检测试剂盒(美国Promega公司),miR-29a mimic、inhibitor(广州锐博生物科技公司)。引物及DNA合成、测序均由上海生工生物工程有限公司完成。主要仪器:高速冷冻离心机(德国Eppendorf公司),PCR仪(美国Applied Biosystems公司),Mini型垂直板电泳系统、硝酸纤维素膜电转系统(美国BIO-RAD公司),GEL LOGIC凝胶分析系统(美国KODAK公司),EPICS XL-4流式细胞仪(美国Beckman Coulter公司),多功能荧光酶标仪(瑞士TECAN公司)。
1.2 实验方法
1.2.1 细胞培养 PC3细胞株由三峡大学肿瘤微环境与免疫治疗湖北省重点实验室保存。细胞使用RPMI 1640培养液,同时加入10%小牛血清、100 U/mL青霉素、100 μg/mL链霉素,置37 ℃,5% CO2培养箱中培养。
1.2.2 质粒构建 携带HPV16 E7基因的重组质粒pcDNA3.1(-)-E7构建方法同前期文献报道[4]。报告基因质粒pGL3-cM-CDK6-CS的构建如下:通过TargetScan软件查找miR-29a种子序列与CDK6 mRNA的3′UTR潜在结合位点,设计以下寡脱氧核酸序列:5′-CGCGTCAGAATGCTCTGTTTTGGTGCTTCAGAATGCTCTGTTTTGG-TGCTTCAGAATGCTCTGTTTTGGTGCTTC-3′;5′-TCGAGAAGCACCAAAACAGAGCATTCTG-AAGCACCAAAACAGAGCATTCTGAAGCACC-AAAACAGAGCATTCTGA-3′(下划线分别为MluⅠ和XhoⅠ粘性末端),由上海生工合成。将2条DNA单链退火后得到双链DNA,命名为“CDK6-CS”,再将CDK6-CS片段克隆到荧光素酶报告基因载体pGL3-cM中。
1.2.3 细胞转染 6孔板内细胞培养到对数生长期,密度约70%~80%为转染最佳状态。转染前1 h,把培养液更换为不含血清及双抗的1640培养液。用培养液将纯化质粒和脂质体LipofectamineTM2000稀释,室温静置20 min后均匀多点加入待转染的培养液,5% CO2、37 ℃恒温培养箱培养4~6 h后更换含10% NBCS的RPMI 1640培养液。研究HPV16 E7功能是通过用HPV16 E7及其载体质粒处理PC3细胞来实现的,实验分2组:pcDNA3.1(-)载体组(PC3-3.1组)和pcDNA3.1(-)-E7组(PC3-E7组);研究miR-29a的功能通过加入miR-29a mimic及inhibitor完成。
1.2.4 Western blot检测蛋白表达 收集对数生长期细胞,提取处理后的细胞总蛋白,采用BCA法测定蛋白浓度后配平,取8 μg总蛋白与5×上样Buffer混匀后煮沸5 min制样,再同5 μL marker分别加入泳道,用10%SDS-PAGE分离胶进行电泳、转膜、封闭,洗涤后加入一抗(鼠抗人CDK6抗体,1∶1000)中室温孵育2 h后,漂洗3次,加入二抗(羊抗鼠IgG-HRP抗体,1∶3000)中室温孵育1 h后,TBST漂洗3次,暗室ECL显影。用凝胶成像系统对X线片中显出的目的条带进行灰度分析,β-actin作为内参照。
1.2.5 半定量RT-PCR检测 转染的细胞培养48 h后用Trizol提取RNA并反转录得到cDNA,再以cDNA为模板,加入引物(PCR引物序列见表1),PCR检测其表达水平。取PCR产物8 μL在2% Agrose凝胶中电泳,利用凝胶成像系统对条带进行灰度分析定量,以β-actin作为内参基因。
1.2.6 荧光素酶活性检测 将报告基因质粒pGL3-cM-CDK6-CS分别瞬时转染细胞PC3-3.1和细胞PC3-E7;同时将报告基因质粒pGL3-cM-CDK6-CS与miR-29a inhibitors瞬时共转染PC3-3.1细胞,或与miR-29a mimics瞬时共转染PC3-E7细胞。转染36 h后收集细胞,加入裂解液100 μL,4 ℃下充分裂解细胞,然后取20 μL至不透光96孔板,加入100 μL荧光素酶底物混匀后置于多功能酶标仪中检测荧光强度。通过荧光素酶活性的高低判断对目的基因的转录调控情况。
1.2.7 流式细胞术检测细胞周期分布 收集对数生长期细胞及上清至15 mL试管,2000 r/min室温离心5 min,弃上清,加1 mL流式固定液重悬细胞,4 ℃固定30 min。加入500 μL流式打孔液重悬细胞并转移至流式管,加0.5 mg/mL PI染料90 μL,室温避光染色30 min,尼龙膜过滤后上机检测。
1.3 统计学方法
用SPSS 18.0统计软件对数据进行统计分析。计量资料以“均值±标准差”表示,组间均数比较采用t检验,以P<0.05为差异具有统计学意义。
2 结果
2.1 HPV16 E7对细胞周期相关基因及miR-29a潜在靶基因的影响
应用半定量RT-PCR检测PC3-3.1和PC3-E7两种细胞中细胞周期相关基因CDK4、CyclinE2、E2F1及miR-29a潜在靶基因CDK6及HBP1的表达情况,发现在PC3-E7中这些基因表达均呈不同程度上调,与PC3-3.1比较,差异均具有统计学意义(均P<0.01),其中CDK6表达差异最大,见图1A、1B。Western blot结果显示PC3-E7细胞中CDK6蛋白表达明显高于PC3-3.1细胞(P<0.01),见图1C、1D。
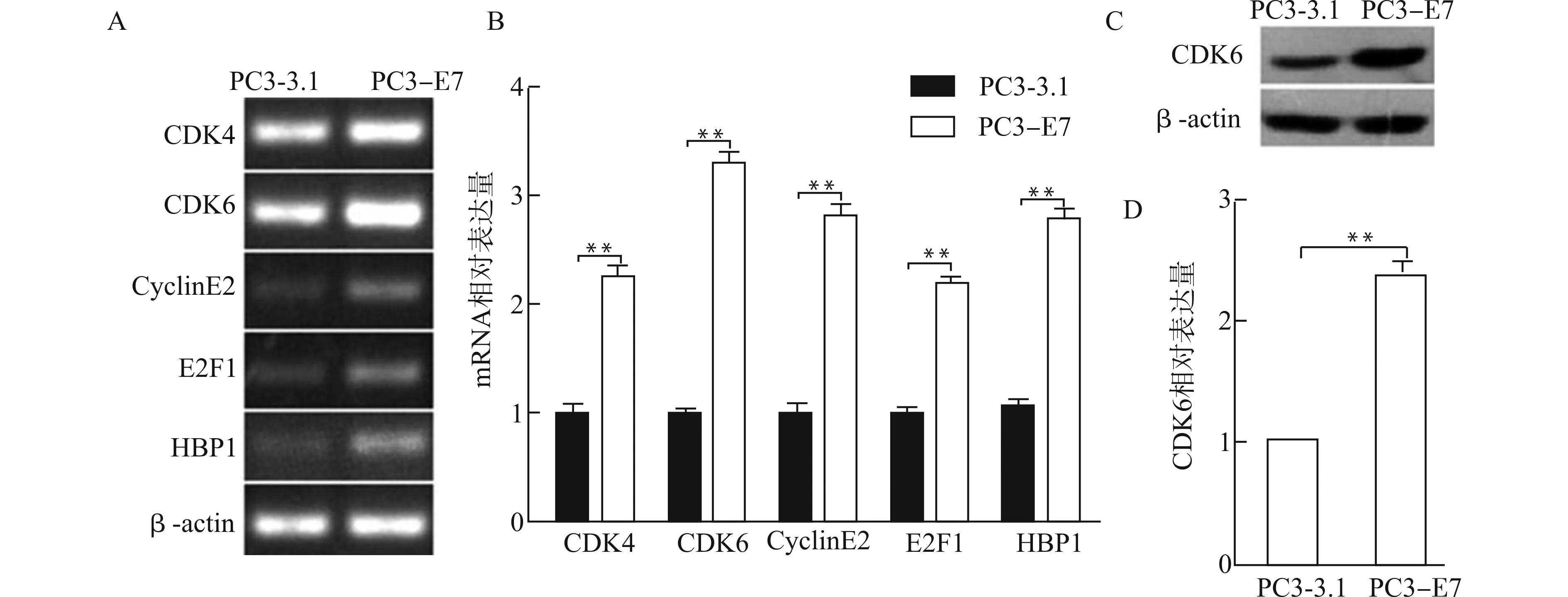
A、B:半定量RT-PCR分析各基因mRNA相对表达量;C、D:Western blot分析CDK6蛋白相对表达量;**P<0.01图1 HPV16 E7对细胞周期相关基因、miR-29a潜在靶基因及CDK6蛋白表达的影响Fig.1 Effects of HPV16 E7 on cell cycle related genes,miR-29a potential target gene and CDK6 protein expression
2.2 miR-29a直接抑制靶基因CDK6的表达
通过TargetScan软件查找miR-29a种子序列与CDK6 mRNA的3′UTR潜在结合位点,见图2A。荧光素酶实验进一步验证发现:与空白对照组相比,miR-29a mimic共转染HPV16 E7组酶活性较低,而inhibitor共转染组酶活性较高,直接证实了miR-29a可与CDK6 3′UTR结合并下调荧光素酶表达;另外PC3-E7细胞荧光素酶活性较PC3-3.1高,见图2B,提示HPV16 E7可以通过下调miR-29a从而促进CDK6表达。
将miR-29a mimics、inhibitors分别瞬时转染PC3-E7和PC3-3.1细胞后,Western blot检测CDK6蛋白的表达,发现转染miR-29a mimic的PC3-E7细胞中CDK6蛋白的表达水平降低(P<0.01,见图2C、2D);而转染miR-29a inhibitor的PC3-3.1细胞中CDK6蛋白的表达水平升高(P<0.05,见图2E、2F)。这进一步证实了HPV16 E7可以通过下调miR-29a而促进CDK6表达。
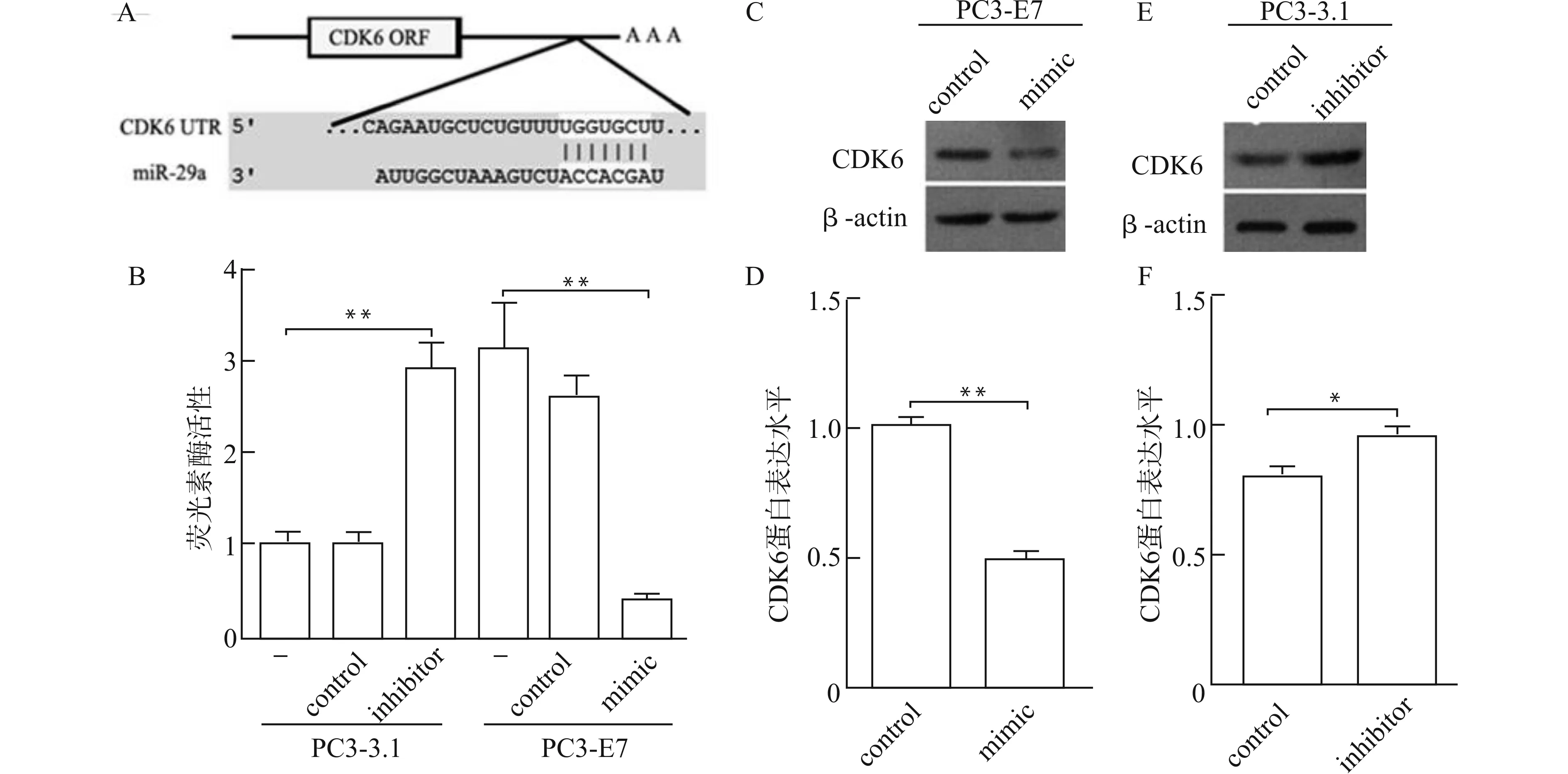
A:预测miR-29a与CDK6 mRNA的3′UTR潜在结合位点;B:荧光素酶报告基因实验结果;C、D:PC3-E7细胞转染miR-29a mimic后Western blot分析CDK6蛋白表达;E、F:PC3-3.1细胞转染miR-29a inhibitor后Western blot分析CDK6蛋白表达;*P<0.05,**P<0.01图2 miR-29a与CDK6的靶向关系Fig.2 Targting relationship between miR-29a and CDK6
2.3 miR-29a对细胞周期分布的影响
将miR-29a mimic和inhibitor分别转染PC3-E7和PC3-3.1,流式细胞术分析细胞周期分布情况,结果如图3所示:向PC3-3.1转染miR-29a inhibitor,G0/G1期细胞减少,而S、G2/M期细胞增多,说明促进细胞周期从G1向S期进展;向PC3-E7细胞转染miR-29a mimic,G0/G1期细胞增多,而S、G2/M期细胞减少。这提示miR-29a通过下调CDK6抑制细胞增殖,恢复PC3-E7细胞中miR-29a表达至少可以部分逆转HPV16 E7的促增殖效应。

A:流式细胞术检测miR-29a对细胞周期分布的影响;B:PC3-3.1细胞转染miR-29a inhibitor后细胞周期中各时期细胞百分比的变化;C:PC3-E7细胞转染miR-29a mimic后细胞周期中各时期细胞百分比的变化;**P<0.01图3 miR-29a对细胞周期分布的影响Fig.3 The effect of miR-29a on cell cycle distribution in cells
3 讨论
宫颈癌是全球范围内最常见的妇科恶性肿瘤之一,在绝大部分的宫颈癌患者中均存在HPV感染。高危型HPV的持续感染是其发病过程中的主要危险因素,其中HPV16和HPV18是最常见的2种致癌亚型。HPV基因组的调控功能主要由E区基因编码,其中E6基因编码的E6蛋白可以调节癌基因p53的表达,发挥致癌作用,而E7基因编码的蛋白可以降解Rb,释放E2F转录因子,从而影响细胞周期的调控,诱导细胞恶性增殖[2,5]。miRNAs是一类长度约为18~25 nt的内源性非编码小RNA,在生物体内起重要的转录后调控作用,广泛参与了细胞的各种生物学行为,在肿瘤的发生发展过程中起到重要的调控作用[6]。众多研究均提示:高危型HPV的E6、E7等致癌基因可以通过影响miRNAs的表达来调控相应靶基因的表达从而促进宫颈癌的发生发展。Kong等[7]研究发现HPV E7蛋白过表达后可以上调miR-21的表达,从而促进HeLa细胞的增殖和侵袭。而抑制E7的表达后HeLa细胞中的miR-21表达下调,并抑制细胞的增殖和侵袭。Chen等[8]研究发现在宫颈癌中高危HPV E7通过TGF-β/Smad4信号通路上调miR-182的表达,其具体机制是高危HPV E7与pRb结合后,E2F从复合物中释放出来并结合到TGF-β启动子区域,导致TGF-β过表达,TGF-β过表达时Smad4信号通路被激活,导致Smad4与miR-182启动子区相互作用,进而导致miR-182在宫颈癌细胞及周围细胞中表达上调,促进宫颈癌的发生和发展。在我们的前期研究中发现HPV16 E7可以促进细胞增殖,加速细胞周期G1向S期进展,其机制与下调miR-29a的表达有关[4]。
miR-29a在包括宫颈癌在内的多种恶性肿瘤中均呈低表达,被认为是一种抑癌基因,其与肿瘤的发生发展密切相关。Nan等[9]研究发现在宫颈癌组织中miR-29a呈低表达,且其表达的下调与宫颈癌不良预后密切相关,进一步研究发现miR-29a通过直接靶向SIRT1参与宫颈癌的迁移、侵袭和上皮间充质转化(EMT)过程,故可以认为miR-29a/SIRT1轴与宫颈癌的发病密切相关。Gong等[10]发现在宫颈癌中miR-29a能够直接抑制DNMT1,抑制SOCS1启动子的甲基化并上调其表达。此外,miR-29a还可以促进宫颈癌细胞凋亡、抑制细胞增殖,并通过抑制NF-κB信号通路来抑制宫颈癌细胞的迁移和入侵,因此推测miR-29a-DNMT1-SOCS1轴可能通过NF-κB信号通路来影响宫颈癌的浸润和转移。Chen等[11]发现宫颈癌细胞中miR-29a表达下调,CDC42上调。荧光素酶报告基因实验显示miR-29a通过直接靶向CDC42的3′UTR负调控其表达。过表达miR-29a后HeLa和CaSki细胞中的PAK1、p-PAK1、p-LIMK和p-cofilin的水平显著下调,细胞凋亡明显增加,而细胞增殖、迁移和侵袭受到明显抑制,而过表达CDC42后可以部分恢复这些变化。Chen等[12]发现miR-29a-3p对SNIP1的mRNA和蛋白表达水平进行负调控,并且下调SNIP1下游基因(HSP27、c-Myc、Cyclin D1)的mRNA表达从而抑制了HeLa细胞的迁移和增殖,认为miR-29a-3p/SNIP1轴可以为宫颈癌的发展提供新的认识。在本研究中,我们通过荧光素酶实验发现miR-29a可以直接靶向CDK6的3′UTR,负调控CDK6的表达,并促进PC3细胞的增殖过程,同样起到抑癌基因的作用,与其他研究中的结论相似,但作用靶点不同。
CDK6与Cyclin D密切相关,二者可形成复合物而促使Rb蛋白发生磷酸化并失活,促进细胞从G1期进入S期。CDK6在大多数肿瘤中呈高表达,具有抑制肿瘤细胞凋亡、促进肿瘤血管生成等作用,与肿瘤进展密切相关[13]。有研究报道,CDK6在宫颈癌组织中呈高表达,且其表达水平与宫颈癌的临床分期、分化程度、淋巴结转移、HPV感染呈显著的正相关性[14]。本研究发现,HPV16 E7阳性细胞中CDK6表达上调,从而促进细胞的增殖。而转染miR-29a mimic可以显著降低CDK6蛋白表达水平,提示miR-29a高表达对肿瘤细胞中致癌基因CDK6表达具有抑制作用,并抑制细胞的增殖。
综上所述,HPV16 E7可以下调肿瘤细胞中miR-29a的表达水平,而miR-29a可通过直接抑制CDK6的表达,进一步促进细胞增殖和抑制其凋亡。这一结果提示E7-miR-29a-CDK6信号可能是高危型HPV致使细胞发生恶性增殖新的重要途径,而恢复miR-29a的表达至少可以部分逆转E7的促增殖效应,这为肿瘤的分子治疗提供了新的靶点。
猜你喜欢
皮肤性病诊疗学杂志(2021年5期)2021-11-27
中学生物学(2021年8期)2021-11-02
天津医科大学学报(2021年4期)2021-08-21
广东蚕业(2021年1期)2021-03-18
三农资讯半月报(2021年1期)2021-01-27
绿色科技(2019年18期)2019-11-22
江苏农业科学(2019年23期)2019-03-03
中国医药导报(2016年33期)2017-03-06
医学信息(2016年29期)2016-11-28
现代养生·下半月(2015年8期)2015-11-16
