
利用全外显子组测序技术对妊娠中期胎儿双肾缺如的病因分析*
2022-01-16 10:34刘海意
华中科技大学学报(医学版) 2021年6期
何 登, 刘海意
华中科技大学同济医学院附属同济医院妇产科,武汉 430030
肾脏缺如是一种临床罕见的先天性出生缺陷,其中胎儿双肾缺如畸形则更为罕见,遗传是其主要致病因素,但发病机制尚不明确。双肾缺如胎儿往往表现为孕中晚期羊水缺失或羊水过少,通过产前B超或MRI检查而被发现。近年来,随着高通量测序技术的发展,全外显子测序(whole exome sequenceing,WES)等技术逐渐被广泛运用于妇产及遗传学科领域,为肾缺如的产前诊断方法提供了新的选择,也为探索其遗传病因及相关机制提供了科学手段。
基因Jagged1(JAG1)变异可致常染色体显性遗传疾病Alagille综合征1型(Alagille syndrome 1),主要表现为:肾发育不良、膀胱输尿管返流、集合管系统重复、局灶节段性肾小球硬化、肋骨异常、肝功能衰竭、室间隔缺损、房间隔缺陷、法洛四联征、主动脉缩窄、肾动脉狭窄、肾小管性酸中毒等。本研究中,我们对1例经彩超检查发现的双肾缺如胎儿,通过全外显子组测序检测为其产前诊断提供了相应依据,发现此前未曾报道的JAG1基因第16号外显子杂合缺失可能参与了胎儿肾缺如畸形的组织学发生。
1 资料与方法
1.1 临床资料
该家系于2020年9月24日因“孕19+6周,彩超检查发现胎儿双肾缺如2 d”收入华中科技大学同济医学院附属同济医院产科。患者因原发不孕于我院生殖中心行ET术开始本次妊娠,2020年9月7日彩超检查提示:双顶径4.0 cm,头围15.1 cm,腹围13.2 cm,股骨长2.3 cm,胎儿相当于孕16+4周;胎盘附着于子宫底部及后壁,羊水最大前后径0.9 cm。初步诊断:羊水过少,胎儿心脏大血管内径异常。予以营养支持及改善胎盘循环治疗,并于2020年9月23日复查胎儿彩超提示:胎儿双侧肾脏未见明显回声,无羊水,胎儿孕周相当于18周;同时胎儿心脏彩超提示胎儿室间隔缺损可能(胎儿室间隔上部0.11 cm回声连续性中断),静脉导管PI值增高(1.30)。胎儿父母非近亲结婚,无不良药物或环境接触史。既往史、家族史无特殊。患者入院完整诊断:胎儿畸形(Potter综合征?室间隔缺损?),羊水过少,妊娠合并糖尿病,ET术后,孕19+6周,G1P0。经遗传咨询后,胎儿父母要求终止妊娠行病因学检查,并签署知情同意书。本研究经过华中科技大学同济医学院附属同济医院伦理委员会批准。
1.2 检测方法
1.2.1 标本采集及DNA提取 经过利凡诺羊膜腔注射引产后,取引产儿腹股沟股内侧直径2.5 cm肌肉组织及引产胎儿父母外周血5 mL,应用北京天根生化科技有限公司的DNA提取试剂盒提取全基因组DNA,并保存于4 ℃环境。
1.2.2 文库构建及全外显子组测序 经过片段化、末端修复、3′段加A尾,连接测序接头、文库片段筛选及PCR扩增到DNA文库。应用Agilent Sureselect Human All Exon V6试剂盒进行全外显子杂交捕获,对于杂交获得的外显子文库进行清洗和纯化,获得富集的外显子DNA文库。PCR扩增后进行磁珠纯化,得到待上机的DNA文库。用Agilent BiosystemsTM7500 Real time PCR systems仪器平台进行qPCR文库定量,之后上机测序。illumina测序平台PE150测序策略为样本的测序条件。最后得到下机数据用于生物信息学分析。
1.2.3 全外显子测序分析 对生成的Fastq格式的原始文件进行评估,并过滤掉Q30碱基未达到80%以上的数据。将通过质控的数据用Burrow-Wheeler Aligner(BWA,Version 0.7.12)比对人类参考基因组,用SAM tools(Version 1.3)将比对后的文件转换为BAM格式,Genome Analysis Toolkit(GATK,Version 3.3)对其进行变异检测。Annovar对变异进行注释,再根据注释信息对变异进行如下过滤:①过滤掉变异碱基型深度低于5×(即A0<5)的变异,以减少由测序误差导致的变异;②过滤掉东亚人群数据库中AF> 0.05的变异(优先级:ExAC东亚人群> 1000 Genomes东亚人群);③筛选出非良性变异;④过滤掉内含子、基因间区中的变异;⑤过滤掉同义变异;⑥过滤掉在本地外显子数据库中AF>0.05的变异;⑦过滤掉隐性遗传病基因中单一的杂合变异。
1.2.4 生物信息学分析方法及数据库 变异位点致病性评级及数据解读规则参考美国医学遗传学与基因组学学会(ACMG)指南及ClinGen序列变异解释(Sequence Variant Interpretation,SVI)专家组对指南标准的应用建议[1-3],排除千人基因组、人类外显子数据库、人群基因组突变频率数据库、神州基因组数据库等数据库中突变频率大于1%的变异位点,去除非功能性变异位点(如同义突变、非编码区突变等),再经过致病性预测(SIFT、Polyphen2、CADD等软件)、临床症状对照、相关疾病数据库查询与文献参考等综合考虑,找到候选基因变异位点进行家系验证。所用注释数据库为:Human Genome 38(hg38/GRCh38)、RefSeq、dbSNP、1000 Genomes phase3、ExAC、gnomAD、神州基因组数据库等数据库,所用解读数据库包括DGV、DECIPHER、OMIM、UCSC、ClinVar、HGMD及PubMed等数据库。
2 结果
2.1 引产胎儿产前彩超及引产后大体观
引产儿产前彩超检查提示胎儿双肾缺如异常,引产后大体观可见胎儿面容鼻梁稍宽符合Alagille综合征的面部畸形表现,同时观察双上臂未见明显异常,双下肢未见明显残缺畸形等情况(图1)。
2.2 全外显子组测序分析结果
通过全外显子组测序对引产儿组织及父母血液样本检测,发现引产儿JAG1基因16号外显子疑似杂合性缺失,父亲和母亲均未发生缺失(图2)。全外显子组测序-染色体数目变异(WES-CNV)结果显示引产儿变异染色体位置位于chr20:10645355-10645502,区段为20p12.2。经公共数据库查询,基因JAG1(OMIM:601920)的突变会导致常染色体显性遗传疾病Alagille综合征1型。
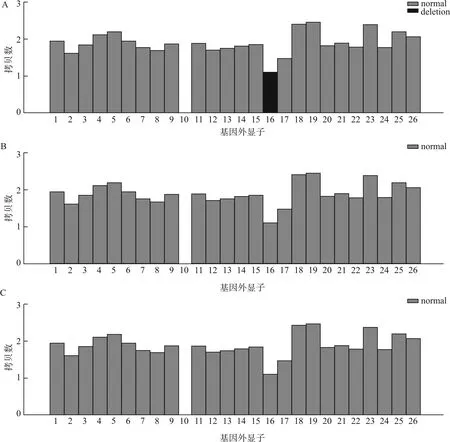
A:受检引产胎儿JAG1基因第16号外显子杂合缺失;B:引产儿父亲未见JAG1基因16号外显子缺失;C:引产儿母亲未见JAG1基因16号外显子缺失图2 全外显子组测序检测范围内染色体数目变异结果Fig.2 CNV results within WES detection
受检者样本未检测到与本案例表型相关的致病性SNV、In Dels变异,不过检测到1个重点关注位点:受检引产标本及父系均为EYA1基因c.361G>C杂合突变,而母系为野生型。根据ACMG指南及ClinGen序列变异解释(Sequence Variant Interpretation,SVI)专家组对指南标准的应用建议[1-3],基因EYA1的c.361G>C突变为一个意义未明突变位点,与常染色体显性遗传疾病“鳃-耳-肾综合征1型伴或不伴白内障”相关。其位于EYA1基因6号外显子c.361G>C:p.A121P。该变异发生于Eyes absent homologue 1功能结构域,疾病特征与本案例表型相符度为部分相符,在人群基因组突变频率数据库(gnomAD)中的频率为5.43951E-05。该变异在神州基因组数据库、人类外显子数据库(ExAC)和参考人群千人基因组(1000G)中没有发现。
本研究对原始标本行全基因组检测同时发现3号染色体q21.3处重复0.48 Mb区域,经查询DGV、DECIPHER、OMIM、ClinGen、UCSC、gnomAD以及PubMed公共数据库资源,未找到与该片段相关的明确致病信息和文献报道(图3)。

A:全基因检测结果;B:3号染色体检测结果图3 全基因组检测同时发现3号染色体q21.3处重复0.48 Mb区域Fig.3 The repeat 0.48 Mb region of chromosome 3 q21.3 found by genome-wide detection
3 讨论
Jagged1(JAG1)基因突变或缺失是目前已知的常染色体显性遗传病Alagille综合征(20p11缺失综合征)(MIM #118450)的常见致病原因[4]。JAGl有大于36 kb的26个外显子,可编码组成1218个氨基酸的Jaggedl蛋白,后者是Notch信号途径中的重要配体。JAGl基因突变或缺失使其编码的蛋白能力下降,从而引起Notch信号传导途径异常,进而造成胚胎细胞、组织、器官的分化和发育异常[5]。文献报道JAGl基因突变在全部26个外显子上均有发现,没有明显引起Alagille综合征的热点位置[6]。本研究收集双肾缺如引产胎儿组织及胎儿父母外周血,对受检者样本进行全外显子组测序CNV检测(WES-CNV),发现JAG1基因16号外显子疑似杂合缺失。
经公共数据库查询,基因JAG1(OMIM:601920)的突变会导致常染色体显性遗传疾病Alagille综合征1型(OMIM:118450)。Alagille综合征1型主要表现为:肾发育不良、膀胱输尿管返流、集合管系统重复、局灶节段性肾小球硬化、肋骨异常、肝功能衰竭、室间隔缺损、房间隔缺陷、法洛四联征、主动脉缩窄、肾动脉狭窄、肾小管性酸中毒、半椎体、尺骨发育低下、不完全外显、婴儿期发病、表现度差异。关于JAG1突变引起Alagille综合征肾缺如病例比较少见,较早文献报道1例Alagille综合征患者经产前检测发现存在多囊性肾发育不良的表现[7]。结合引产胎儿彩超提示双肾缺如结果、胎儿父母泌尿系彩超结果及WES-CNV结果显示父亲和母亲均未发生缺失,考虑本病例可能为胎儿父母双方JAG1杂合突变引起胎儿JAG1基因杂合缺失可能。
此外,在本研究中受检者样本未检测到与本病例表型相关的致病性SNV、In Del变异。已有研究发现了80多种位于8q13.3染色体的EYA1基因的突变可引起鳃-耳-肾综合征[8-10]。鳃-耳-肾综合征(OMIM #113650、#610896)是一种常染色体显性遗传病,其表现包括鳃裂缺陷(存在侧颈部瘘管或囊肿)、耳凹、听力损失以及肾脏异常,包括肾脏未发育和发育不全。鳃-耳-肾综合征的发病率为1/40000。鳃-耳-肾综合征患者的外显度可能不全,可能仅具备一个或两个特征[11]。并非所有患者都会出现肾异常,同一家族中不同成员的肾脏异常可能不同。肾脏表现包括肾缺如或发育不全(单侧或双侧)、VUR、UPJO或输尿管重复畸形[12]。EYA1基因负责编码肾发育期间在后肾间充质中表达的一种转录辅因子[13]。EYA1基因与SIX基因家族成员相互作用,后者负责编码控制后肾间充质中PAX2和GDNF蛋白产物表达的转录因子。鳃-耳-肾综合征家族中还存在SIX1基因(位于14q23.1染色体)和SIX5基因的突变。本研究发现基因EYA1的c.361G>C突变与常染色体显性遗传疾病鳃-耳-肾综合征1型(Branchiootorenal syndrome 1)相关。考虑为意义未明突变位点可能,即EYA1∶NM_000503.6∶exon6∶c.361G>C∶p.A121P变异。该变异发生于Eyes absent homologue 1功能结构域,而且变异在神州基因组数据库、人类外显子数据库(ExAC)和参考人群千人基因组(1000G)中没有发现,在人群基因组突变频率数据库(gnomAD)中的频率为5.43951E-05。EYA1(OMIM∶601653)的突变会导致临床上鳃-耳-肾综合征1型发生,表现为:肾发育不良、羊水过少、膀胱输尿管返流、肾缺如、多囊性肾发育不良、外耳道狭窄、小耳畸形、耳蜗发育不全、鳃裂瘘管、鳃裂囊肿、表现度差异、不完全外显。结合患病胎儿羊水过少及双肾缺如病理改变,EYA1基因变异可能与本病例发病相关。
综上,本研究发现了中孕期双肾缺如胎儿病变可能与JAG1基因16号外显子杂合缺失密切相关,此外,基因EYA1的c.361G>C突变为一个意义未明突变位点,亦可能与胎儿双肾缺如相关。本研究也存在一定局限性,由于双肾缺如胎儿病例极为罕见,后续将进一步搜集病例和验证我们的新发现结果。目前全外显子组检测已成熟应用于遗传咨询,然而也存在一定局限:①全外显子组检测主要针对全外显子组区域及剪切边界5 bp范围内的单碱基变异(SNVs)和外显子区域50 bp以内的插入/缺失(In Dels),不包含poly结构、串联重复序列、富含GC区域以及存在同源相似序列(假基因),同时对50 bp以上的插入/缺失(In Dels)存在一定的局限性;②全外显子组捕获的技术局限性,目前全外显子组检测不能保证覆盖100%的外显子区域;③多基因病及其他非遗传因素(感染、药物、辐射等环境因素)引起的可能性。本研究首次报道JAG1基因第16号外显子杂合缺失可能与胎儿双肾缺如为临床特征的Alagille综合征1型疾病相关,本发现对遗传咨询领域相关资料提供了一定程度的补充。
猜你喜欢
分子诊断与治疗杂志(2022年9期)2022-10-09
军事文摘(2022年16期)2022-08-24
今日农业(2022年4期)2022-06-01
今日农业(2021年14期)2021-10-14
今日农业(2021年11期)2021-08-13
科学之谜(2019年3期)2019-03-28
科学之谜(2018年8期)2018-09-29
中学生理科应试(2016年4期)2016-11-19
恋爱婚姻家庭·养生版(2016年9期)2016-09-07
湖北农业科学(2014年11期)2014-09-10
