
ERK抑制剂增强多柔比星诱导乳腺癌细胞凋亡的作用
2021-10-14 08:58钱江华刘亚明杜家如
中国药理学通报 2021年10期
钱江华,刘亚明,杜家如,刘 浩
(蚌埠医学院药学系,安徽 蚌埠 233000)
内质网(endoplasmic reticulum,ER)是真核细胞中负责蛋白质合成、修饰、折叠和组装的场所,在缺氧、糖基化抑制、氧化应激等因素作用下,内质网腔未折叠蛋白或错误折叠蛋白聚集,引起ER应激(ER stress)[1]。为增强对蛋白的折叠和清除能力,减少内质网应激的损害,恢复内质网蛋白稳态,未折叠蛋白反应(unfolded protein response,UPR)被激活[2-3]。UPR由3种跨膜蛋白启动,分别是活化转录因子6(activating transcription factor 6,ATF6)、肌醇需求酶1 (inositol-requiring enzyme 1,IRE1)和蛋白激酶样内质网激酶(protein kinase-1ike ER kinase,PERK)。生理状态下3条信号通路蛋白(ATF6、IRE1、PERK)与葡萄糖调节蛋白78(glucose-regulated protein78,GRP78)结合,处于非活性状态。在ER应激作用下,GRP78与以上3种跨膜蛋白分离,并结合错误折叠蛋白,从而激活UPR信号通路。
MAPK 通路的异常活化会导致细胞丧失分化和凋亡的能力,引起细胞异常增殖、转化,在肿瘤耐药方面发挥重要作用[4]。研究表明,多数实体肿瘤对线粒体介导凋亡敏感性下降的一个重要原因是MAPK/MEK/ERK信号通路的激活,调节MAPK信号通路是缓解临床多药耐药的一个重要途径[5]。
多柔比星(doxorubicin,adriamycin,ADM,阿霉素)为蒽环类抗生素,通过阻止RNA转录过程,抑制RNA合成广泛用于恶性淋巴肉瘤、乳腺癌、卵巢癌等恶性肿瘤的治疗[6-8]。但是由于心脏毒性及耐药性,一定程度上限制了其在临床中应用。
本研究以临床治疗乳腺癌常用的药物多柔比星和抑制糖基化的物质衣霉素(Tunicamycin,TM)作用乳腺癌细胞,观察乳腺癌细胞对多柔比星引起ER应激途径的细胞凋亡反应及抑制MEK通路引起细胞凋亡的改变,为探讨减少多柔比星耐药及提高疗效提供新思路。
1 材料
1.1 细胞株乳腺癌细胞株MDA-MB-231细胞(购自中科院细胞所)。
1.2 试剂DMEM 高糖培养基(批号11995500)、DMEM 低糖培养基(批号11885500)、胰蛋白酶(批号25200072):Gibco公司;胎牛血清(批号 90090531):杭州四季青公司;GRP78(批号sc-166490)、ERK1(批号sc-271270)、ERK2(批号sc-271451)、pERK1/2(批号sc-135900)及β-actin(批号sc-58673):SantaCruz公司;山羊抗兔IgG(批号20000217)、山羊抗小鼠IgG(批号20000242):中杉金桥生物技术有限公司;TM(批号654380)、PD98059(批号167869):Sigma公司;多柔比星,规格:每支10 mg,批号:19059911,批准文号:H33021980,海正瑞辉制药有限公司生产。
2 方法
2.1 细胞培养人乳腺癌细胞MDA-MB-231加入DMEM高糖型培养基,37 ℃、5% CO2饱和湿度环境下培养传代。
2.2 MTT法测定细胞增殖活性取对数生长期的MDA-MB-231细胞以每孔100 μL培养液含6 000个细胞密度接种于96孔板中,在37 ℃、5% CO2培养箱中培养24 h后弃去培养液,用不同浓度(0、1、2、4、8、12 μmol·L-1)TM和(0、0.25、0.5、0.75、1.0、1.5 μmol·L-1)ADM处理,每组设3个复孔。继续培养24、48、72 h后每孔加入浓度为5 g·L-1的MTT溶液10 μL继续孵育4 h,弃上清;每孔加DMSO150 μL,37 ℃孵育30 min,酶标仪测定570 nm波长吸光度A值,重复实验3次。
2.3 Western blot检测相关蛋白表达MEK抑制剂PD98059(40 μmol·L-1)预先处理MDA-MB-231细胞1 h后再给予TM(4 μmol·L-1)及ADM(0.5 μmol·L-1)处理不同时间(0、6、12、24、36 h),每个时间点处理24 h后,提取总蛋白,BCA法测定各组蛋白浓度。SDS-PAG电泳,转膜,无蛋白快速封闭液室温封闭30 min,加入一抗(GRP78、ERK1/2、pERK1/2,稀释比例均为1 ∶1 000)4 ℃过夜。TPBS洗涤3次,二抗1 ∶10 000室温孵2 h,TPBS洗涤3次。最后使用ECL系统对蛋白的表达情况进行检测。
2.4 PI染色检测细胞凋亡将MDA-MB-231制成单细胞悬液,以每孔1×105个细胞数接种于24孔细胞培养板,培养24 h后加入PD98059(40 μmol·L-1)、ADM(0.5 μmol·L-1)及PD98059联合ADM处理,继续培养48 h后收集培养液至流式管中,1 200 r·min-1离心10 min,弃上清;培养板中加入PI 缓冲液750 μL/孔,37 ℃孵育10 min,收集细胞于对应流式管中,混匀,4 ℃避光保存,过夜,流式细胞仪检测。以亚G1期DNA含量的细胞比例代表凋亡细胞数。
2.5 RT-PCR检测GRP78转录水平根据GRP78基因的全长序列设计引物:GRP78上游:5′-GTTTGCTGAGGAAGACAAAAAGCTC-3′,下游:5′-CACTTCCATAGAGTTTGCTGATAATTG-3′,扩增长度为240 bp。取5 μL PCR产物与1 μL 6×上样缓冲液混合,上样于2.0%琼脂糖凝胶,以1×TBE缓冲液电泳,电压100 V,电泳30 min。电泳图谱进行灰度分析,求得样品电泳条带灰度与内参照GAPDH条带灰度之比,进行半定量。

3 结果
3.1 TM和ADM对MDA-MB-231细胞增殖抑制作用MTT结果表明,随着TM和ADM浓度的增加和作用时间的延长,细胞的存活率下降。如Fig1示,4 μmol·L-1的TM作用于MDA-MB-231 细胞后24、48、72 h,细胞存活率分别是(92.72±3.98)%、(90.13±3.65)%和(82.69±4.47)%。0.5 μmol·L-1的ADM作用于 MDA-MB-231 细胞后24、48、72 h,细胞存活率分别是(90.35±3.37)%、(60.77±4.31)% 和(54.27±4.97)%。
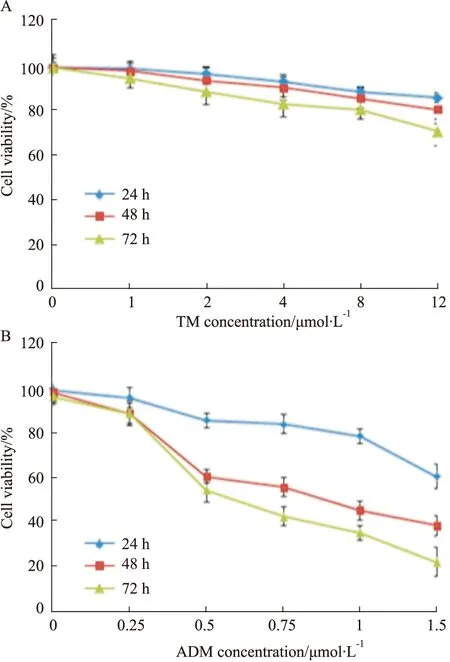
Fig 1 Changes of viability of MDA-MB-231 cells after treatment with TM and ADM for 24,48 and 72 h
3.2 GRP78在乳腺癌组织及细胞中的表达
3.2.1GRP78在乳腺癌组织中呈阳性表达 免疫组化实验结果显示,GRP78在乳腺癌组织中表达与乳腺正常组织中表达有差异(P<0.05),GRP78在乳腺癌组织中强阳性表达,而在乳腺正常组织中呈弱阳性表达,见Fig2。
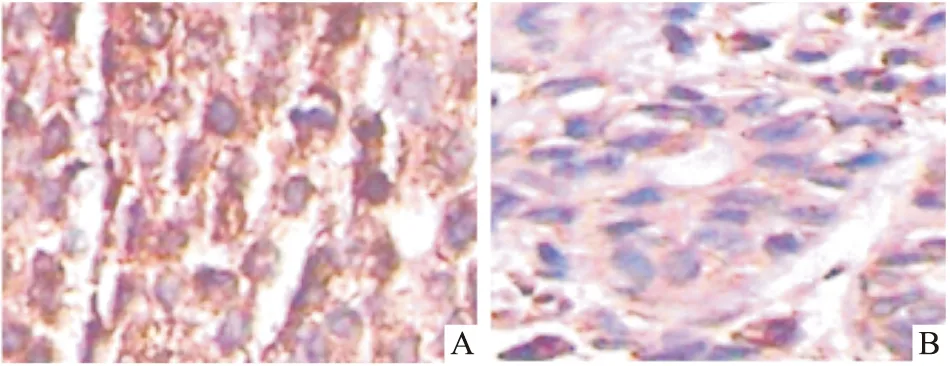
Fig 2 Expression of GRP78 in breast tissues(×400)A:GRP78 is strong positive in breast cancer;B:GRP78 is weak positive in normal breast
3.2.2TM、ADM上调MDA-MB-231细胞中GRP78的表达 Fig3 Western blot检测结果可以看出,GRP78在MDA-MB-231细胞中(0 h)呈阳性表达,并且TM、ADM均可上调GRP78的表达。TM作用24 h时表达最强,差异有显著性(P<0.01),至36 h时表达减弱。ADM作用36 h时表达最强,差异有显著性(P<0.01)。
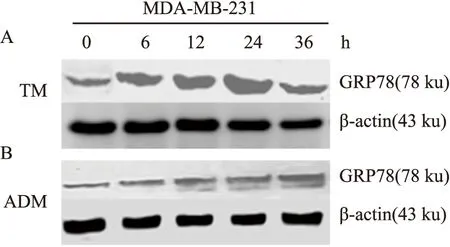
Fig 3 TM and ADM induced activation of GRP78 in MDA-MB-231 cells
3.3 PD98059增强ADM诱导乳腺癌MDA-MB-231细胞的凋亡Fig4A Western blot检测显示,对照组中pERK1/2正常表达,因MEK/ERK通路被阻断,其下游的pERK1/2表达(PD98059组及ADM+PD98059组)显著降低。Fig4B凋亡检测结果显示,PD98059单独作用时细胞凋亡率为17%,ADM单独作用时细胞凋亡率为20%,但二者合用时凋亡率高达45%,与单独作用时比较,差异有显著性(P<0.05)。
3.4 PD98059抑制ADM对GRP78的表达Fig5A Western blot检测显示,PD98059作用于MDA-MB-231细胞不同时间,与对照组(0 h)比较,PD98059下调乳腺癌细胞GRP78的表达。ADM与PD98059联合处理细胞,Fig5B结果显示PD98059可抑制ADM对GRP78的上调作用,不同的时间组GRP78表达与对照组(0 h)比较无明显差异(P>0.05)。
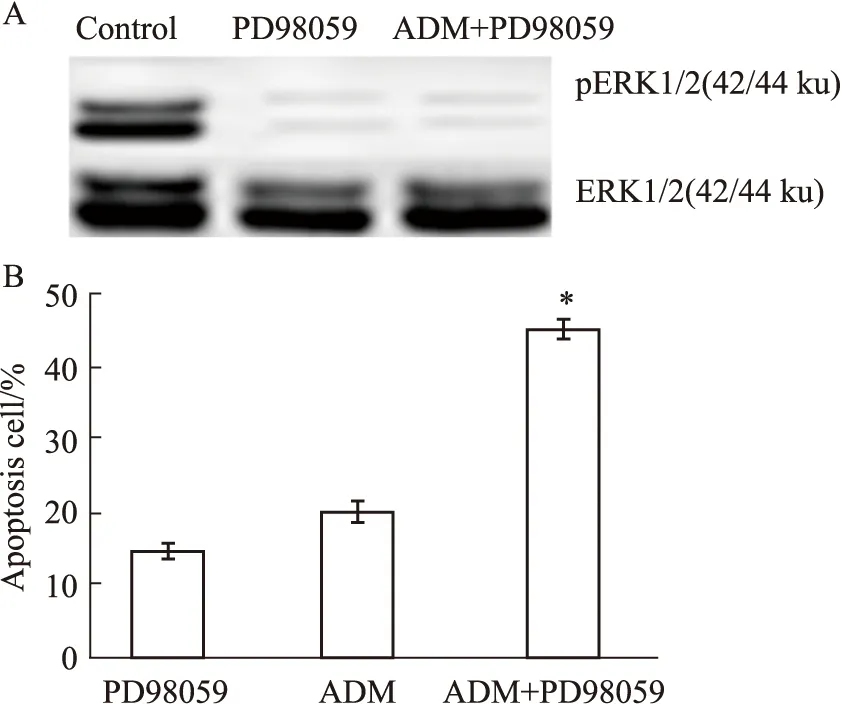
Fig 4 Effects of PD98059 on expression of pERK1/2 and apoptosisA:PD98059 blocked activation of pERK1/2;B:PD98059 sensitized MDA-MB-231 cells to ADM-induced apoptosis.*P<0.05 vs ADM and PD98059 group

Fig 5 Effect of PD98059 on GRP78 expressionA:PD98059 down-regulated GRP78 expression;B:PD98059 blocked ADM-induced up-regulation of GRP78 expression
3.5 抑制MEK/ERK通路对GRP78转录水平的影响如Fig6所示,ADM组GRP78 mRNA表达水平高于对照组(P<0.05),PD98059组GRP78 mRNA表达水平低于对照组(P<0.01),ADM联合PD98059组GRP78 mRNA表达水平低于ADM组和对照组(P<0.01)。
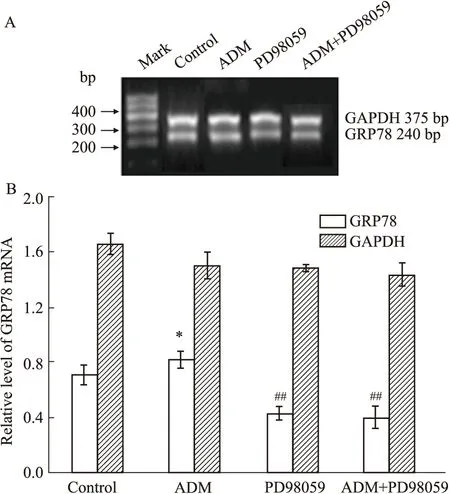
Fig 6 Effect of PD98059 on mGRP78 expressionA:PD98059 decreased GRP78 mRNA expression level;B:Variation of GRP78 mRNA expression level.*P<0.05 vs control group;##P<0.01 vs control and ADM group
4 讨论
乳腺癌依旧是全球女性高发恶性肿瘤之一,常规化疗或内分泌治疗等不仅促进癌细胞演化,获得新的基因组变异[9],也可通过改变细胞的应激反应如氧化应激、内质网应激等,使癌细胞适应不断变化的肿瘤微环境,导致癌细胞对抗癌药物敏感性下降,发生耐药[10-12]。
MEK/ERK信号通路是细胞外信号向细胞内转导的重要途径,也是多种药物抑制肿瘤增殖和转移的重要环节[13]。内质网应激有利于肿瘤细胞获得更强的生存能力[14]。作为UPR的中心调节蛋白 GRP78具有的抗凋亡作用可使肿瘤细胞逃避抗癌药物的杀伤,导致疗效下降。靶向 GRP78诱导凋亡的合成肽 BMTP-78,能选择性的杀死乳腺癌细胞,抑制原发性肿瘤的生长及转移[15]。本实验研究表明,常用的抗肿瘤药物ADM能引起乳腺癌发生ERS,上调乳腺癌细胞GRP78的表达,提示ADM激活UPR引起GRP78高表达是乳腺癌细胞对ADM诱导凋亡不敏感的原因之一。
与单用ADM和PD98059相比,二者合用的凋亡率大大增加,说明抑制MEK通路可以增加乳腺癌细胞对ADM引起内质网应激途径凋亡的敏感性。进一步实验结果表明,PD98059抑制ADM对乳腺癌细胞GRP78 的上调表达,说明抑制ADM对乳腺癌细胞UPR的诱导是PD98059增加乳腺癌细胞对ADM的敏感性原因之一。
可见,UPR信号通路和MEK/ERK通路的激活是肿瘤耐药性产生的2个重要环节。有研究结果指出UPR信号通路和MEK/ERK通路可能是抑制肿瘤转移和治疗肿瘤的重要靶点[16-17],但UPR在肿瘤耐药、转移方面的机制仍需更为深入的研究。本研究提示阻断MEK/ERK通路的激活可增加乳腺癌细胞对ADM诱导凋亡的敏感性,这一作用与抑制ADM对乳腺癌细胞UPR的诱导有关,为ADM在肿瘤增殖、细胞耐药的分子机制研究提供新的思路。
猜你喜欢
航天电子对抗(2022年4期)2022-10-24
解放军医学杂志(2021年12期)2022-01-18
现代临床医学(2021年1期)2021-01-26
中国科技纵横(2018年2期)2018-11-29
中成药(2018年7期)2018-08-04
安徽医科大学学报(2016年12期)2017-01-15
中国药物应用与监测(2015年5期)2015-12-11
中国当代医药(2015年33期)2015-03-01
西南军医(2014年4期)2014-01-19
中国医疗器械杂志(2013年5期)2013-12-05
