
液相色谱-串联质谱法测定水果中吡虫啉的不确定度评定
2021-09-01 13:33:14◎赵飞
现代食品 2021年12期
◎ 赵 飞
(辽宁省检验检测认证中心,辽宁 沈阳 110000)
不确定度作为评价分析结果准确性的定量表征,其数值越小,表明结果准确性越高[1]。测量不确定度的评定是根据采用的检测方法对待测组分进行检测所得到的检测数据评价的一种方式,也是评价检测方法合理性、科学性的有效手段之一[2]。
吡虫啉通过作用于昆虫的乙酰胆碱受体,扰乱神经活动达到杀虫效果,具有触杀和胃毒双重作用,应用较为广泛[3]。由于吡虫啉具有较强的内吸作用,不当使用后可能造成农药残留超标,给人体健康带来安全隐患。此外,吡虫啉对蜜蜂等传粉生物具有较大毒性,可严重抑制蜂群数量的增长,间接影响环境生态平衡[4]。目前,吡虫啉的检测多采用高效液相色谱法和高效液相色谱-串联质谱法[5]。《食品安全国家标准 食品中涕灭砜威、吡唑醚菌酯、嘧菌酯等65种农药残留量的测定 液相色谱-质谱/质谱法》(GB 23200.34—2016)中规定的方法可一次处理检测多种农残,本文按照该标准进行水果中吡虫啉的检测,并对检测过程的不确定度进行系统的分析,为检验检测实验室按照该方法开展水果中吡虫啉检测提供科学的指导与依据。
1 材料和方法
1.1 仪器与试剂
Waters XEVO TQD液相色谱-串联质谱仪(美国沃特世公司);BSA224S-CW电子天平(德国赛多利斯公司);GM200高速粉碎机(德国莱驰公司);FJ300-SH均质器(德国IKA公司);X1R高速冷冻离心机(德国Thermo公司)。
水果购于当地市场。丙酮、二氯甲烷、乙腈(色谱纯,美国Fisher公司);无水硫酸钠、氯化钠(分析纯,国药集团化学试剂有限公司);甲酸(色谱纯,上海安谱公司);吡虫啉标准物质(纯度为98.5%,Dr. Ehrenstorfer)。
1.2 方法
1.2.1 标准曲线配制
准确称取吡虫啉标准品20.52 mg于20 mL容量瓶中,用乙腈定容至刻度,即得浓度为1.0 mg·mL-1的标准储备液。准确吸取0.1 mL浓度为1.0 mg·mL-1的标准储备液用乙腈稀释定容至100 mL,即得1.0 µg·mL-1吡虫啉标准中间液。基质标准系列溶液:准确吸取标准中间液(1.0 µg·mL-1)0.25 mL、0.50 mL、1.0 mL、2.5 mL和5.0 mL于50 mL容量瓶中,用5%乙腈定容至刻度,配成标准系列工作液。准确移取以上各浓度标准系列工作液1.0 mL分别添加至经空白样品前处理的残渣中,涡旋混合均匀,制得5.0 ng·mL-1、10 ng·mL-1、20 ng·mL-1、50 ng·mL-1和100 ng·mL-1基质标准系列溶液,临用现配。
1.2.2 仪器条件
色谱柱:WATERS BEH C18,2.1 mm×50 mm;流速:0.4 mL·min-1;柱温:30 ℃;进样量:5 µL。梯度洗脱程序:A为乙腈,B为0.1%甲酸水;0~0.5 min,A:5%;0.5~2.5 min,A:5%~95%;2.5~3.5 min,A:95%~5%。离子源温度:150 ℃;检测方式:MRM;毛细管电压:2.0 kV;脱溶剂气流量:800 L·h-1;脱溶剂气温度:400 ℃;定量离子255.9/174.8;定性离子255.9/208.9;锥孔电压:34 V;碰撞电压:12 V/16 V。
1.2.3 样品制备
称取10 g试样,加10 mL水,静置;再加40 mL丙酮,振荡提取;上述溶液减压抽滤,滤液合并浓缩。向浓缩液中加入氯化钠溶液和二氯甲烷各30 mL,振荡,移取二氯甲烷层;再向剩余溶液中加入30 mL二氯甲烷,重复上述操作,合并二氯甲烷层。二氯甲烷层经无水硫酸钠脱水,浓缩至近干,加入2.0 mL SPE溶液溶解。将上述溶液转入固相萃取柱(预先采用SPE溶液淋洗),收集流出液;再用SPE溶液洗涤固相萃取柱,收集流出液;流出液浓缩至近干,氮气吹干;再加入一定体积乙腈,再用0.1%甲酸定容至1.0 mL,涡旋混匀,过滤膜待测。
1.2.4 数学模型的建立
建立测定样品中吡虫啉含量的数学模型(1)。

式(1)中,ω-样品中吡虫啉的含量,µg·kg-1;ρ-由标准曲线得出的样液中吡虫啉的浓度,ng·mL-1;ρ0-空白溶液中吡虫啉的浓度,ng·mL-1;V-样品定容体积,mL;c-系数;A-试样溶液中吡虫啉的响应值;As-标准溶液中吡虫啉的响应值;m-水果样品的称样量,g;P-水果空白基质的加标样品回收率。
2 结果与分析
2.1 标准不确定度分量的评定
2.1.1 标准物质引入的不确定度ur(s)
(1)标准物质纯度引入的不确定度ur(c1)。由标准物质证书可知,吡虫啉的相对不确定度U=1.07%(k=2)。由标准物质纯度引入的相对标准不确定度为:

(2)标准溶液配制引入的不确定度ur(c2)。标准品称量采用分度为0.000 01 g天平,称量范围在0~50 g时,其最大允许误差为±0.5 mg,按均匀分布计算,称量操作过程中由使用的分析天平而引入的相对不确定度为:

准确吸取吡虫啉标准储备液0.1 mL于100 mL容量瓶中,用乙腈定容至刻度,即得1.0 µg·mL-1吡虫啉标准中间液。
使用0.1 mL的A级分度吸量管准确吸取0.1 mL吡虫啉标准储备液于100 mL容量瓶中,用乙腈稀释并定容,配制成吡虫啉标准中间液,其质量浓度为1.0 µg·mL-1。该过程的不确定度来源主要由两方面引入,一方面是使用的吸量管、容量瓶等玻璃器皿本身引入的,另一方面是由于操作时实验室的温度发生变化,进而导致量取的溶液体积变化而引入不确定度。0.1 mL的A级分度吸量管允许误差为±0.002 mL,按照均匀分布考虑,则由此引入的不确定度为:

A级100 mL容量瓶的容量允许误差为±0.10 mL,按照均匀分布考虑,取则容量瓶引入的不确定度为:

标准溶液配制室的温度控制在(20±5)℃,20 ℃下乙腈的膨胀系数为1.37×10-3/℃,则由乙腈体积变化的相对标准不确定度为:

综上,标准中间液配制过程引入的不确定度为:
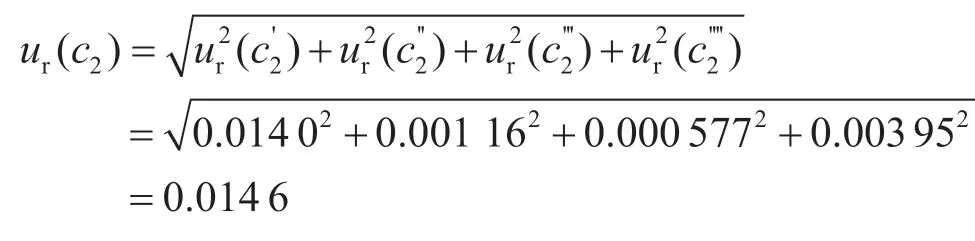
(3)基质标准溶液配制引入的不确定度ur(c3)。由基质标准溶液配制引入的各分量相关情况见表1。

表1 基质标准溶液配制过程引入的分量表
根据各分量,基质标准系列溶液配制过程引入的 相对标准不确定度为:

(4)标准曲线拟合引入的不确定度ur(c4)。本方法采用优化后的仪器条件,分别对5个不同浓度的标准溶液进行测定,浓度依次为5.0 ng·mL-1、10 ng·mL-1、20 ng·mL-1、50 ng·mL-1和100 ng·mL-1。以吡虫啉的浓度为横坐标、其响应峰面积为纵坐标,进行线性拟合,相关数据见表2。向阴性样品中加标,并进行6次平行测定,测得加标样品的测试液浓度分别为:45.6 ng·mL-1、45.2 ng·mL-1、45.0 ng·mL-1、45.3 ng·mL-1、45.7 ng·mL-1和45.4 ng·mL-1,平均值为45.4 ng·mL-1,由标准曲线引入的不确定度可由公式(2)计算。

表2 吡虫啉基质标准曲线表

式(2)中,u(c4)-曲线拟合引入的不确定度;S(A)基质标准溶液中吡虫啉响应值残差的标准差;b-基质标准曲线的斜率;p-同一溶液测量次数(6次);n-基质标准溶液的测定次数(5次);c0-以基质标准曲线校正待测样品浓度的平均值,ng·mL-1;ci-基质标准曲线各点浓度的理论值,ng·mL-1;c-i-基质标准曲线各点浓度的平均值,ng·mL-1。
由基质标准曲线拟合引入的相对标准不确定度为ur(c4)为0.005 42。其中,S(A)可按公式(3)计算。

式(3)中,Ai-基质标准溶液各点的响应值;A-以基质标准曲线计算得到的理论响应值;n-基质标准曲线的点数。
综上,标准物质引入的相对标准不确定度计算公式为:

2.1.2 样品称量引入的不确定度ur(m)
样品称量时采用分度为0.01 g的分析天平。0~50 g称量范围内该天平的最大允许误差为±0.05 g,按均匀分布,由样品称量操作过程引入的相对不确定度为:
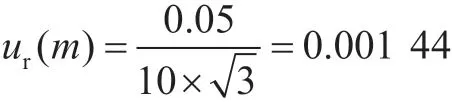
2.1.3 样品前处理引入的不确定度ur(V)
提取过程中净化后加入0.4 mL乙腈,再加入0.6 mL 0.1%甲酸水溶液。分别采用0.5 mL、1 mL分度吸量管进行添加,容量允差分别为±0.005 mL、±0.008 mL,该过程引入的相对标准不确定度为:
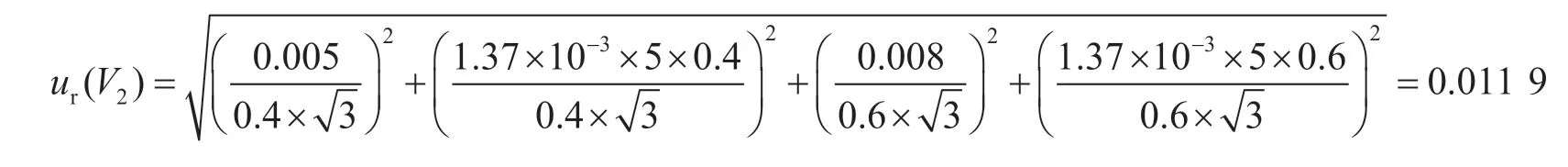
2.1.4 重复性引入的不确定度ur(R)
按照实验方法,对阴性样品进行加标实验,添加量为5 μg·kg-1,测定值分别为:4.56 μg·kg-1、4.52 μg·kg-1、4.50 μg·kg-1、4.53 μg·kg-1、4.57 μg·kg-1和4.54 μg·kg-1,加标测试样品的平均测定值为4.54 μg·kg-1,采用贝塞尔公式计算待测物的实际标准偏差为0.005 69,则相对标准不确定度为:
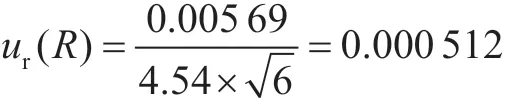
2.1.5 回收率引入的不确定度ur(P)
对阴性水果样品进行加标实验,添加量吡虫啉的量相当于5 μg·kg-1,回收率分别为:91.2%、90.0%、90.6%、91.4%、90.8%和90.4%,平均值为90.7%,按照A类评定应用贝塞尔公式计算待测物的实际标准偏差为0.569%,其标准不确定度为:

采用t检验方法来判断本实验的回收率同100%之间是否存在显著差异,若存在,需要采用回收率对结果进行修正。当n=6,在95%置信概率水平下t0.95(5)=2.571,该回收率下即t>2.571,即检测结果需采用回收率进行修正。
本实验中,由回收率引入的相对标准不确定度为:
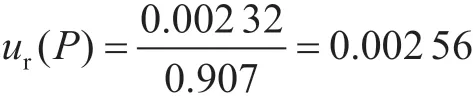
2.2 合成不确定度
综合以上各分量,吡虫啉的合成相对标准不确定度计算如下:
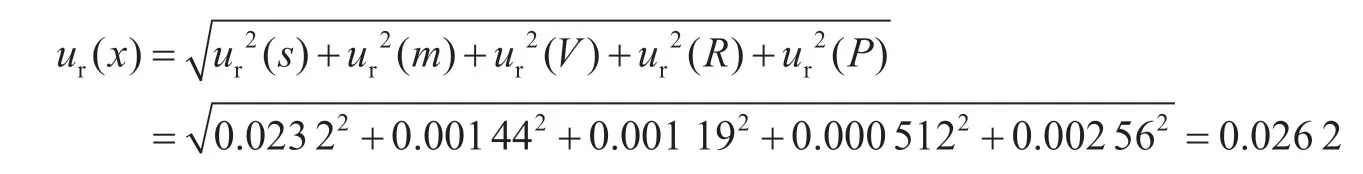
合成标准不确定度:u(x)=0.026 2×4.54=0.12 μg·kg-1。
2.3 扩展不确定度和结果表示
当置信概率在95%时,吡虫啉检测结果的扩展不 确 定 度为U95=k×u(x)=0.24 μg·kg-1(k=2)。按照《测量不确定度评定与表示》(JJF 1059.1—2012)要求,采用本方法检测样品中吡虫啉的含量,检测结果应表示为(4.54±0.24)μg·kg-1。
3 结论
采用液相色谱-串联质谱法测定水果中吡虫啉含量,对整个操作过程中存在的不确定度分量进行系统评定。通过评定可以看出,液相色谱-串联质谱法进行水果中吡虫啉检测,基质标准溶液的配制引入的不确定度贡献最大,回收率和样品称量引入的不确定度贡献次之。因此,今后的工作中,标准系列溶液配制时应通过更合理地设置标准曲线浓度,选用合适的移液工具,严格控制实验室的温度,提高检测人员的技术水平,切实减小不确定度分量,保证检测数据的科学性和合理性。
猜你喜欢
煤化工(2022年3期)2022-07-08 07:24:42
云南化工(2021年9期)2021-12-21 07:43:42
河北果树(2021年4期)2021-12-02 01:14:40
四川蚕业(2021年4期)2021-03-08 02:59:56
化工设计通讯(2020年10期)2020-09-17 14:43:16
世界热带农业信息(2018年3期)2018-09-26 07:56:20
Cancer Biology & Medicine(2016年4期)2017-01-13 01:54:45
中国氯碱(2016年9期)2016-11-16 03:07:39
中国资源综合利用(2016年10期)2016-01-22 08:36:09
化学分析计量(2013年5期)2013-03-11 16:37:43
