
高密度脂蛋白在动脉粥样硬化免疫反应中的作用机制研究进展
2021-07-28 09:39潘杭雨刘丹郎吉萍戴秋月郭志刚
解放军医学杂志 2021年6期
潘杭雨,刘丹,郎吉萍,戴秋月,郭志刚
南方医科大学南方医院心血管内科,广州 510515
心脑血管疾病是人类健康的第一大杀手,而冠状动脉粥样硬化是心血管疾病的首要病因[1]。2016年,全球1/3死亡者的病因是心血管疾病,其中超过75%发生在低收入和中等收入国家,给这些国家带来了巨大的经济负担[2]。降低危险因素、提高保护因素是预防和治疗动脉粥样硬化的重要措施。高密度脂蛋白(high density lipoprotein,HDL)作为一种血清蛋白,一直以来被认为是心血管疾病的重要保护因素之一。流行病学研究表明,血浆低高密度脂蛋白胆固醇(high density lipoprotein-cholesterol,HDL-C)和高低密度脂蛋白胆固醇(low density lipoproteincholesterol,LDL-C)是动脉粥样硬化性心血管疾病(atherosclerotic cardiovascular disease,ASCVD)的主要危险因素[3]。Boden等[4]发现,血浆中HDL-C每升高10 mg/L,心血管疾病的患病风险可降低2%~3%。目前一致认为,胆固醇逆转运(reverse cholesterol transport,RCT),即胆固醇从外周组织及细胞中流出转运至肝脏经胆道排泄是降低ASCVD风险的主要机制,而胆固醇通过HDL从巨噬细胞中流出是RCT的关键步骤[5]。近年来,越来越多的研究提示动脉粥样硬化与免疫反应也存在密切联系。在动脉粥样硬化形成过程中,单核细胞来源的巨噬细胞吞噬脂质后形成泡沫细胞是动脉粥样硬化斑块发生的一个重要环节,而且在斑块病灶中还可发现树突细胞,以及与其共定位的T淋巴细胞。HDL促进胆固醇外流的作用也调控了参与动脉粥样硬化的免疫细胞的一系列反应,包括单核-巨噬细胞、B淋巴细胞和T淋巴细胞等[6]。因此,HDL可通过影响免疫反应而对动脉粥样硬化的发生发展过程产生影响,从而发挥抗动脉粥样硬化的作用。本文以HDL为重点,结合多项文献研究阐述其与免疫反应的相互作用关系,以期为动脉粥样硬化的防治提供新的策略。
1 HDL概述
HDL是脂质与蛋白质的非共价准球形复合物,其水合密度为1063~1210 g/L[7]。HDL颗粒的基本结构包括酯化的胆固醇核心和由磷脂、游离胆固醇(free cholesterol,FC)、载脂蛋白组成的单层结构外壳[8]。HDL颗粒具有不同比例的不同脂质:三酰甘油(triglycerides,TG)、胆固醇酯(cholesterol ester,CE)、FC及磷脂。部分HDL颗粒缺乏脂质或无脂质[9]。随着对HDL研究的深入,鉴定出了几种特定的HDL,约70%的HDL由载脂蛋白A-Ⅰ(ApoA-Ⅰ)组成,而由ApoA-Ⅱ组成者占15%~20%[7]。缺乏脂质的ApoA-Ⅰ主要在肝脏和小肠中合成,分泌入血浆后与三磷酸腺苷结合盒转运体A1(ATPbinding cassette transporter A1,ABCA1)相互作用产生前体β-HDL[10]。ABCA1将细胞内的FC转运至细胞膜,并流出至缺乏脂质的ApoA-Ⅰ形成盘形HDL。卵磷脂胆固醇脂酰转移酶(lecithin-cholesterol acyltransferase,LCAT)可酯化HDL表面的FC,使HDL形态由盘形转变为球形,成为成熟的HDL。在人类H D L 中的C E 可通过胆固醇酯转移蛋白(cholesteryl ester transfer protein,CETP)转移至富含TG的脂蛋白,间接通过LDL受体在肝脏中清除,或直接通过选择性摄取作为HDL中CE肝受体的B类Ⅰ型清道夫受体(scavenger receptor class B type Ⅰ,SR-BⅠ),而在肝脏中清除[9]。
如前所述,HDL最为人所熟知的功能是通过促进胆固醇从巨噬细胞中流出而促进RCT。HDL还具有抗炎、抗氧化、抗凋亡及促进一氧化氮(nitric oxid,NO)产生等作用[11]。一项在冠心病患者中进行的研究发现,稳定型冠心病或急性冠脉综合征(acute coronary syndrome,ACS)患者的HDL无法刺激内皮细胞一氧化氮合酶激活途径及NO的产生,从而导致HDL的内皮抗炎及修复作用受损[12]。因此,HDL促进NO的产生可能在抗动脉粥样硬化进程中发挥了重要作用。此外,胰岛β细胞内脂质水平失衡被认为是胰岛β细胞衰竭的重要原因。HDL可通过降低胰岛β细胞内胆固醇的含量,改善其分泌胰岛素的功能,从而在血糖调节及缓解2型糖尿病中发挥作用[13]。
2 HDL与免疫反应
人体免疫反应主要分为两大部分:获得免疫和固有免疫。获得免疫是机体应对特异性抗原时产生的免疫反应,通过抗原递呈细胞(antigen presenting cell,APC)加工抗原并产生大量的T淋巴细胞或B淋巴细胞受体和免疫球蛋白来识别抗原;固有免疫是出生后即具备的天然防御系统,参与的免疫成分主要有吞噬细胞、NK细胞、溶菌酶和补体等,对外来抗原起到非特异性防御作用。目前已有大量研究证实,获得免疫和固有免疫中的一些成分参与了动脉粥样硬化的发生发展过程,后续内容将主要针对目前已有较多基础或临床研究证明的与HDL和动脉粥样硬化炎症反应相关的免疫机制进行阐述。
2.1 HDL与获得免疫 在动脉粥样硬化斑块中发现的多种APC提示获得性免疫反应可发生于斑块病灶中[14]。大量研究发现,多种获得免疫中的关键因素参与了动脉粥样硬化的发生发展过程,如定位于细胞膜上的脂筏,可促进炎症反应的鞘氨醇-1-磷酸(spinghosine-1-phosphate,S1P),以及在高胆固醇血症小鼠中发现的产生促炎因子γ-干扰素和肿瘤坏死因子(tumor necrosis factor,TNF)的辅助性T细胞1(helper T cell 1,Th1)均可发挥促动脉粥样硬化作用[14-16]。而三磷酸腺苷结合盒转运体G1(ATPbinding cassette transporter G1,ABCG1)及ABCA1在巨噬细胞、树突细胞中表达也证实了HDL和ApoA-Ⅰ可参与APC调节胆固醇内稳态的过程[17]。
2.1.1 HDL与脂筏 脂筏是含有高浓度胆固醇、鞘磷脂和与多种生物学过程相关的蛋白质的细胞膜微结构域,目前认为其参与调节了LDL的转胞吞作用、血管壁细胞的凋亡、免疫细胞的激活与炎症反应、蛋白酶的激活以及斑块的稳定性等一系列与动脉粥样硬化相关的关键事件。定位于APC脂筏中的主要组织相容性复合体Ⅱ(major histocompatibility complex Ⅱ,MHC Ⅱ)类分子,在抗原递呈和信号转导中发挥了重要作用[17]。脂筏能将MHC Ⅱ肽复合物聚集于APC表面,减少活化T细胞所需的抗原量,因此,破坏APC表面的脂筏结构可抑制MHCⅡ介导的低浓度抗原递呈[18]。脂筏的功能与其含有的脂质成分息息相关,增加或减少细胞膜中胆固醇的含量会改变脂筏的结构,进一步导致脂筏依赖的信号通路发生改变[19]。HDL和ApoA-Ⅰ可促进胆固醇从细胞中流出,使脂筏中胆固醇的含量发生变化,导致巨噬细胞及其他APC的功能发生重要改变,并对抗原递呈产生影响,抑制了APC激活T细胞的功能[16-17](图1),而ApoA-Ⅰ与ABCA1结合激活的信号通路在该效应中可能起主要作用[14]。有研究证实,当巨噬细胞、单核细胞、中性粒细胞、内皮细胞和脂肪细胞暴露于HDL或ApoA-Ⅰ时,可导致脂筏丰度降低,从而广泛抑制多种脂筏依赖的炎症反应[20]。CD40及其配体CD40L可激活多条炎症信号通路,从而促进动脉粥样硬化病灶的发生发展。CD40通过募集它的适配蛋白TNF受体相关因子(TNF receptor-associated factor,TRAF)形成蛋白复合物,进一步激活下游促炎信号通路,而TRAF-6在被配体激活后主要定位于脂筏中。ApoA-Ⅰ可通过ABCA1依赖的胆固醇外流调节TRAF-6进入脂筏,从而抑制内皮细胞核因子-κB(nuclear factor kappa-B,NF-κB)的活化,减轻CD40L诱导的巨噬细胞炎症反应[21]。总之,HDL和ApoA-Ⅰ可通过调节胆固醇的外流改变脂筏的功能进而影响免疫细胞,从而对动脉粥样硬化产生影响。
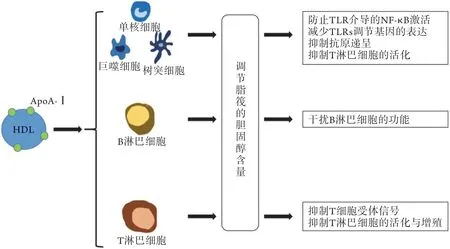
图1 HDL调节脂筏胆固醇含量对抗原递呈细胞功能的影响Fig. 1 Influence of HDL regulating cholesterol content of lipid rafts on the function of antigen presenting cells
2.1.2 HDL与鞘氨醇-1-磷酸 脂筏中的鞘脂产生鞘氨醇,再被鞘氨醇激酶磷酸化后产生S1P。游离的或与白蛋白结合的S1P均容易被降解,而与HDL结合的S1P较稳定,提示HDL-S1P可能是发挥生物学功能的主要形式[16]。在巨噬细胞中,有S1P受体1(S1P receptor 1,S1PR1)和S1P受体2(S1P receptor 2,S1PR2)两种受体,S1P可通过S1PR1抑制脂多糖(lipopolysaccharide,LPS)介导的促炎细胞因子的产生,且可使促炎的M1巨噬细胞亚型向抗炎的M2亚型转变;还可通过S1PR2抑制巨噬细胞的迁移和向炎症部位的聚集[14]。正常情况下S1PR2在内皮细胞中几乎不表达,当发生炎症时,S1PR2的表达增加,且其过表达可激活NF-κB依赖的黏附分子的产生[22],因此S1P在动脉粥样硬化进程中的作用具有两面性:一方面,S1P具有抗细胞凋亡、抗炎、舒张血管、维持内皮细胞屏障功能的作用,且可降低血管细胞黏附分子(VCAM)的表达;另一方面,S1P能够促进淋巴细胞的激活,并促进血栓形成[23]。最近有研究表明,在LDL-R-/-的小鼠中,移植缺乏鞘氨醇激酶2的骨髓可使得内源性S1P水平升高1.5~2.0倍,与野生型小鼠相比,实验组小鼠的病灶绝对大小、坏死核心区域面积和斑块管腔比均明显降低[24]。S1P可能是HDL发挥心血管保护作用的基础,且有研究认为HDL是与载脂蛋白M结合的S1P的主要载体[25]。Lee等[26]发现,HDL相关S1P可通过刺激S1PR1和SR-BⅠ受体蛋白的分子间相互作用来影响S1P介导的细胞代谢改变,如可引起细胞内钙离子浓度的升高。Keul等[22]发现,在冠状动脉疾病患者中,由于S1P缺陷可使HDL-S1P水平较健康人低20%(P<0.05),导致HDL功能障碍进而引起NO依赖的动脉血管舒张功能受损;而在健康人中,HDL能够依赖HDL-S1P有效地抑制TNF-α介导的炎症反应。除冠状动脉疾病外,HDL-S1P含量减少还可导致一系列疾病,如2型糖尿病、慢性肾病,提示HDL-S1P可作为HDL的一种重要组分影响HDL的正常功能,并可在疾病中发生改变[22]。
2.2 HDL与固有免疫 蛋白质组学分析显示,HDL颗粒存在56种蛋白质,包括载脂蛋白和免疫调节蛋白,因此HDL可能参与了固有免疫并介导抗炎和抗感染效应[27-28]。HDL可抑制内皮细胞黏附分子的表达,包括细胞间黏附分子-1、VCAM-1及E-选择素。此外,HDL还可抑制促炎细胞因子和化学因子的表达[29-30]。Taborda等[31]分析了HDL对胆固醇结晶和其他天然免疫激活物的免疫调节活性的影响,发现HDL能够在不影响免疫细胞表面模式识别受体表达的情况下,通过Toll样受体(Toll-like receptor,TLR)和炎症小体介导对天然免疫刺激化合物的免疫抑制作用,导致白细胞介素-1β(Interleukin-1β,IL-1β)和IL-6的产生减少,提示HDL可调节不同天然免疫激活物诱导的免疫应答,从而影响炎症反应。炎症小体、炎症反应中产生的急相蛋白及外源性病原体的产物等均可诱导炎性因子的产生,进而引发机体固有免疫反应,并对免疫细胞产生影响,促进动脉粥样硬化病变的发展。HDL可抑制与动脉粥样硬化有关的炎症相关成分,从而发挥抗动脉粥样硬化作用。
2.2.1 HDL与核苷酸寡聚化结构域(NOD)样受体(NLR)蛋白3炎症小体 NLR是炎症反应的重要组成部分,目前共发现了5种NLR炎症小体,其中NOD样受体蛋白3(NOD-like receptor protein 3,NLRP3)炎症小体与多种自身炎症疾病相关,包括痛风和动脉粥样硬化[31]。NLRP3炎症小体可能是连接脂蛋白代谢异常与动脉粥样硬化的关键环节。抑制巨噬细胞中NLRP3炎症小体的关键成分可完全阻滞胆固醇结晶介导的IL-1β的产生[32]。氧化型低密度脂蛋白(ox-LDL)、氧化型高密度脂蛋白(ox-HDL)和HDL对NLRP3及其下游[如含半胱氨酸的天冬氨酸蛋白水解酶-1(caspase-1)、IL-1β、IL-18]的表达具有调控作用,其中ox-LDL和ox-HDL可增加NLRP3的表达,并激活NLRP3下游细胞因子和caspase-1,进一步诱导IL-1β和IL-18的分泌;而在HDL的作用下,NLRP3及其下游炎性细胞因子的表达降低[32]。Thacker等[33]发现,用重组HDL制剂(reconstituted HDL,rHDL)处理单核细胞和原代巨噬细胞可降低胆固醇结晶诱导的IL-1β的产生(P<0.01);分别用rHDL和天然HDL处理单核细胞,与未经处理的对照组相比,IL-1β的分泌分别降低70%和60%(P<0.001),提示rHDL和天然HDL均可通过减少IL-1β和NLRP3等炎症小体关键基因的表达而发挥对IL-1β分泌的抑制效应。由于HDL和ApoA-Ⅰ分别通过ABCG1和ABCA1介导胆固醇的外流,因此在缺乏ABCA1或ABCG1的树突细胞中,ApoA-Ⅰ和HDL介导的胆固醇外流减少,导致胆固醇积累的小鼠表现出NLRP3炎症小体激活,但其活化机制尚未确定[34]。上述研究正向或反向地说明了HDL可减少NLRP3的表达与激活,并进一步抑制炎性因子的产生及炎症反应。
2.2.2 HDL与血清淀粉样蛋白 血清淀粉样蛋白(serum amyloid A,SAA)是急性时相血清中HDL的载脂蛋白[35]。在急性炎症反应中,SAA急剧升高,而在动脉粥样硬化等慢性炎症中,其水平在一定程度上也高于基线水平。在炎症过程中,肝脏产生的急性时相蛋白可取代HDL中的ApoA-Ⅰ、ApoA-Ⅱ和其他酶等,产生富含SAA和血清磷脂酶A2(serum phospholipase A2,sPLA2)的缺乏保护作用的功能障碍型HDL[16](图2)。SAA激活的巨噬细胞刺激IL-8和单核细胞趋化蛋白-1的释放,其功能是分别募集中性粒细胞和单核细胞至炎症部位。其他一些已知与动脉粥样硬化相关的细胞因子的分泌也增加,而HDL可减弱此炎症效应,可能是由于隔绝了具有促炎效应的不与脂质结合的SAA[36-37]。在心脏组织中,SAA诱导VCAM-1、NF-κB、TNF-α和促凝组织因子的mRNA表达明显上调,而在动脉粥样硬化模型ApoE-/-小鼠中,与单用SAA处理的小鼠比较,使用HDL预处理的小鼠血管动脉粥样硬化和脂质氧化损伤程度更轻,表明HDL可降低SAA介导的血管内皮损伤[38]。SAA可通过NLRP3介导的caspase-1刺激巨噬细胞分泌IL-1β,而HDL能够抑制巨噬细胞中SAA介导的IL-1β的转录和分泌[39]。目前关于SAA对HDL功能的影响尚存在争议,有学者认为,在炎症时HDL与SAA的相互作用增强,可导致HDL与脂肪细胞表面的蛋白多糖结合,从而限制HDL与细胞膜的相互作用,并抑制其促进胆固醇外流的能力[40];而另有研究报道,在SAA水平较高的患者中,HDL具有更强的抗氧化能力[41]。总之,SAA作为一种急性时相蛋白在促进动脉粥样硬化病变的慢性炎症中发挥了一定的作用,而HDL能够在一定程度上减轻其带来的炎症效应。
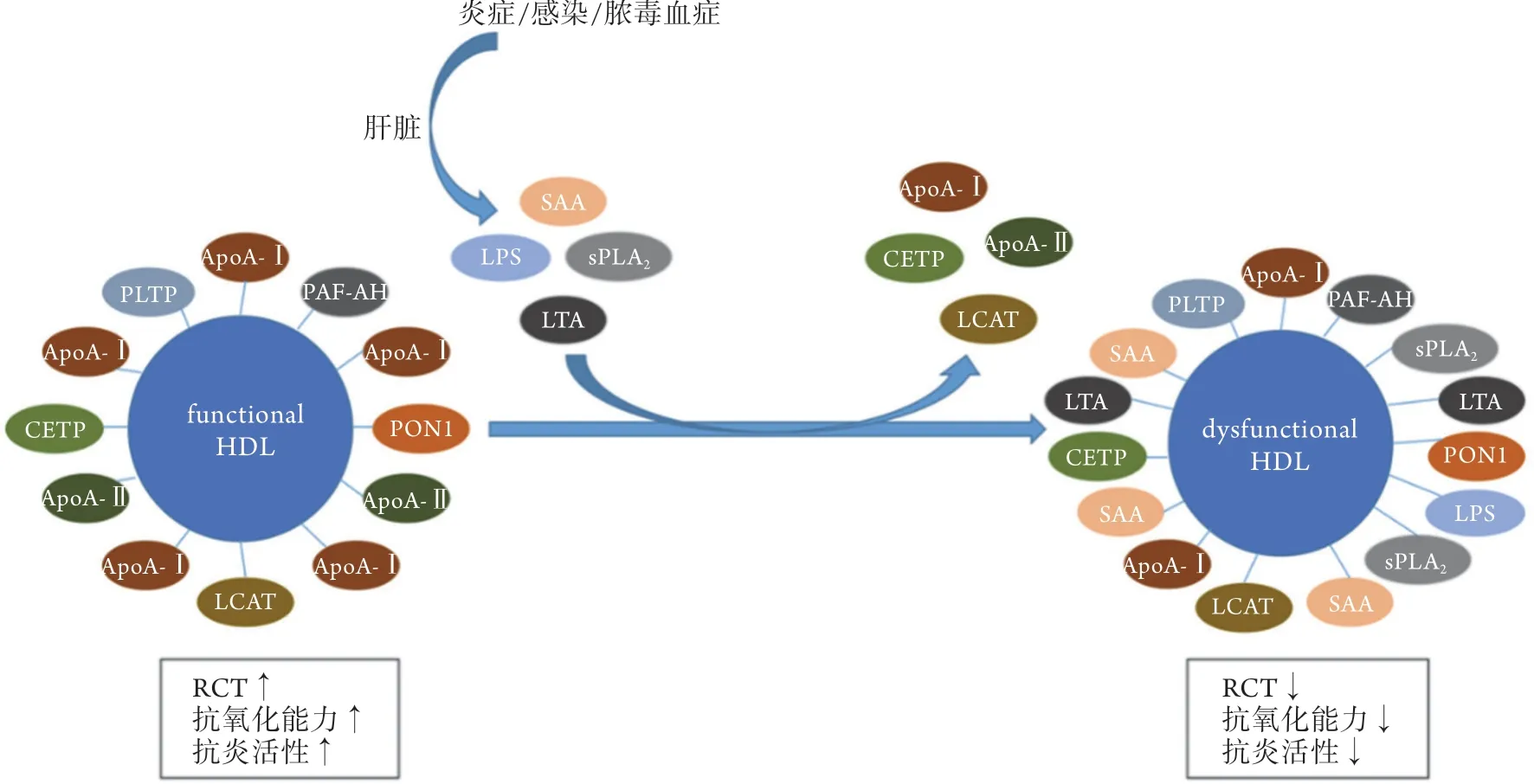
图2 功能型HDL向功能障碍型HDL的转变机制Fig.2 Mechanism of transition from functional HDL to dysfunctional HDL
2.2.3 HDL与LPS LPS通过分泌衰老相关因子(如NF-κB)介导的炎症反应与多种老年退行性疾病密切相关[42]。研究发现,LPS可诱导衰老巨噬细胞的产生,而衰老巨噬细胞可沉积于血管内皮下,并促进弹性纤维的降解,使斑块纤维帽变薄,降低其稳定性,最终促进了动脉粥样硬化病变的发展[43-44]。HDL能够与LPS和其他细菌产物结合并中和毒力,是HDL抑制炎症反应的一个主要机制。HDL可促进LPS与LPS结合蛋白(LPS-binding protein,LBP)的相互作用,有助于LPS的清除;高浓度LBP可抑制白细胞对LPS的反应,而HDL可抑制高浓度LBP的抑制活性,维持白细胞对LPS的敏感性,从而诱导对革兰阴性菌的早期炎症反应[45]。另外,HDL可促进HDL受体SR-BⅠ与HDL-LPS复合物结合并介导复合物的清除[14,45]。有学者在小鼠体内预注射rHDL使其血浆HDL-C水平升高2倍,然后再给小鼠注射LPS,结果显示,HDL-C升高小鼠的血浆TNF-α浓度-时间曲线下面积为(194±132) U/(ml.h),仅为对照组小鼠[(1126±582) U/(ml.h)]的17%(P<0.03),且存活时间明显延长(P<0.05)[46]。相反,在ApoA-Ⅰ敲除小鼠的脓毒血症模型中,由于HDL-C水平较低,中和LPS毒性的能力受损,从而促进了炎性细胞因子的产生[14]。与HDL紧密相连的ApoA-Ⅰ主要通过上调一种能够促进炎症基因mRNA衰减的RNA结合蛋白——tristetraprolin而抑制巨噬细胞中LPS诱导的炎症反应[21]。TLR在动脉粥样硬化病变过程中可启动NLRP3炎症小体,使后者被胆固醇结晶激活,产生IL-1β[47]。LPS是TLR4的一个主要配体,由于TLR定位在脂筏中,因此,当HDL和ApoA-Ⅰ引起脂筏的结构及功能发生改变时,可抑制LPS介导的细胞活化[6]。
3 HDL作为动脉粥样硬化的治疗靶点
目前,治疗动脉粥样硬化的经典药物主要有他汀类、贝特类、烟酸等,近几年也出现了一些针对新靶点的药物如CETP抑制剂、过氧化物酶体增殖物激活受体激动剂、内皮酯酶抑制剂、LCAT调节子等[3]。HDL是降低ASCVD患病风险的主要因素之一,且具有调节免疫的作用,因此,将HDL作为ASCVD的治疗靶点也是研究的一大热点。考虑到HDL的保护作用,如提高循环中的HDL含量可增强其促进RCT及免疫调节的功能,从而减轻动脉粥样硬化中的免疫反应。如前所述,HDL可通过细胞膜上的脂筏实现对免疫细胞的调节,因此,当HDL含量增加时,由于脂筏的作用可抑制免疫细胞的活化,从而减轻炎症反应。
静脉输注HDL是最符合人体生理的一种直接升高HDL水平的治疗方法。rHDL是来源于ApoA-Ⅰ并从血浆中纯化得到的试剂,不仅可以快速升高循环中HDL的水平,而且其中的ApoA-Ⅰ还能够与不同的磷脂重组,而这些磷脂可能产生特定的生物学作用[48]。ApoA-Ⅰ Milano(AIM)是目前知名度最高的rHDL,是载脂蛋白的一种变体,最早在米兰乡村的一小部分人群中发现。2006年,Nicholls等[49]纳入47例30~75岁的ACS患者,随机分组接受安慰剂(n=11)与AIM(n=36)治疗,并使用血管内超声检测血管变化,结果发现AIM组外弹性膜体积与基线相比缩小4.8%[基线时为(343.8±92.5)×10-6L,随访时为(324.5±97.3)×10-6L,P=0.006],粥瘤的体积缩小10.9%[基线时为(163.4±54.3)×10-6L,随访时为(145.6±46.9)×10-6L,P<0.0001],且二者呈正相关关系(r=0.62,P<0.0001),但由于粥样硬化的消退伴随着外弹性膜的逆向重构,导致管腔大小无明显改变[基线时为(180.1±68.3)×10-6L,随访时为(178.9±76.6)×10-6L,P=0.59]。另一种rHDL——CSL111是由人血浆ApoA-Ⅰ和大豆磷脂酰胆碱结合而成,在化学和生物学上与天然HDL相似[50]。Tardif等[50]通过随机对照研究纳入183例患者,并使用血管内超声评估冠状动脉粥样硬化斑块,发现短期的CSL111输注并未明显降低动脉粥样硬化斑块体积(安慰剂组为158.3×10-6L,实验组为151.0×10-6L,P=0.48),但斑块特征指数及冠状动脉积分却明显改善(P=0.01,P=0.03)。近年来,CSL111的替代物——CSL112的临床Ⅱa期试验结果表明,稳定的ASCVD患者单次输注CSL112具有明确的安全性,并能够增强RCT早期阶段关键生物标志物的表达[48]。在一项纳入44例动脉粥样硬化疾病患者的随机双盲对照试验中,CSL112与标准双联抗血小板疗法联用对血小板的聚集无明显影响,因此认为CSL112不会影响急性心肌梗死患者标准双联抗血小板治疗的止血效果或增加心肌梗死后亚急性期出血的风险[51]。
然而,并非所有临床研究都能得出理想的阳性结果。Nicholls等[52]开展的MILANO-PILOT研究对122例使用他汀类药物治疗的ACS患者每周静脉注射MDCO-216(一种新型HDL类似物)或生理盐水安慰剂,6周后通过血管内超声评估动脉粥样硬化斑块情况。结果发现,MDCO-216治疗后斑块减少0.21%,而对照组斑块减少0.94%(P=0.07,95%CI -0.07~1.52);MDCO-216组标准化总动脉粥瘤体积减少6.4×10-6L,而对照组减少7.9×10-6L(P=0.67,95%CI -5.6~8.7)。从检测的各项指标可以看出,在他汀药治疗的背景下,MDCO-216并不能明显增加动脉粥样硬化斑块的消退。该团队的另一项研究(CARAT研究),通过对比CER-001(含有重组人ApoA-Ⅰ与鞘磷脂的rHDL制剂)与安慰剂在ACS患者中的疗效,发现CER-001并未减轻动脉粥样硬化病变[53]。因此,对于不同种类rHDL制剂的效果应当保持中立客观的态度。
4 总结与展望
目前已有大量研究证实,HDL对动脉粥样硬化的发生发展起着重要的负向调节作用,其促进RCT、抗氧化、抗血栓形成等效应均为降低ASCVD患病风险的作用机制,而HDL对免疫炎症反应的抑制是其中的一个重要部分。目前的动物及体外实验表明HDL可通过多种机制,如影响获得免疫中的脂筏、S1P等,以及影响固有免疫中的NLRP3、SAA、LPS等来发挥其抗炎作用,并显示出较好的效果。但目前仍缺乏通过这些机制发挥作用的临床药物,因此,将基础研究真正运用于临床也是研究的另一大方向。同时,对于HDL的免疫调节作用有许多方面仍处于探索阶段,需要开展更多的研究加以阐明。
猜你喜欢
今日农业(2022年14期)2022-09-15
保健医苑(2022年1期)2022-08-30
中老年保健(2022年3期)2022-08-24
中老年保健(2022年4期)2022-08-22
自我保健(2021年2期)2021-11-30
妇女之友(2021年9期)2021-09-26
昆明医科大学学报(2020年11期)2020-12-28
天津医科大学学报(2019年6期)2019-08-13
百姓生活(2019年2期)2019-03-20
制造技术与机床(2017年6期)2018-01-19
