
毕赤酵母醇氧化酶2启动子上游调控序列的随机突变研究
2021-06-30 00:59胡诗艺熊紫莹周化岚张建国
工业微生物 2021年3期
刘 冰,胡诗艺,熊紫莹,韦 雪,周化岚,张建国
上海理工大学 医疗器械与食品学院 食品科学与工程研究所,上海 200093
毕赤酵母(Komagataellaphaffii,曾命名Pichiapastoris) 是能够利用甲醇作为唯一碳源生长的甲基营养型微生物,是目前一种重要的表达外源蛋白的工业微生物[1]。其过氧化酶体中的醇氧化酶1(AOX1)和醇氧化酶2(AOX2)将甲醇氧化为甲醛。醇氧化酶对甲醇和氧气的亲和力比较低[2,3],因此,需要大量的醇氧化酶来代谢甲醇。但是PAOX1和PAOX2都受甲醇严格调控,调控机制相同[4,5]。毕赤酵母在含有葡萄糖、乙醇、甘油等碳源的培养基中生长时,PAOX1和PAOX2都被抑制,细胞中无法检测到AOX1和AOX2的mRNA[2,6-7]。PAOX1和PAOX2在甲醇为唯一碳源的培养基中表达。由于醇氧化酶2启动子(PAOX2)比醇氧化酶1启动子(PAOX1)弱,导致AOX2的含量只占总醇氧化酶量的5%[6,7]。AOX1的基因序列(aox1)和AOX2的基因序列(aox2)高度同源,但它们的启动子区域的DNA序列没有同源性。醇氧化酶启动子强度的差异是由醇氧化酶启动子上游转录调控序列控制,若将aox2置于PAOX1控制下,重组菌株具有AOX1相似的活力。相反,若将aox1置于PAOX2控制下,AOX1的表达量显著下降[6]。目前PAOX1因其调控严格,表达能力强等原因而被广泛研究[8-14],但是依然没有明确其调控序列。而PAOX2的上游调控序列相关信息更少[15,16]。因此,突变PAOX2这样的弱启动子会比强启动子PAOX1更容易实现外源蛋白表达量的升高[17]。而且,研究PAOX2的上游调控序列较易得到相关调控序列,有利于阐明醇氧化酶启动子的调控序列。OHI 等[15]对毕赤酵母PAOX2上游调控序列进行敲除和插入序列,鉴定出PAOX2的一个上游激活序列(UAS)和两个上游抑制序列(URS)。其中UAS与PAOX1的UAS相似。另外,戴秀玉[18]等从aox1基因缺陷菌株Muts中筛选得到Mut+表型突变体,经过测序后发现PAOX2的突变增强了aox2基因的转录,因此可以通过改变PAOX2的上游调控区域来改善aox1基因缺陷菌株利用甲醇的能力,使其在基因工程药物表达及产业化方面发挥重要作用。
本研究以GH11家族的黑曲霉木聚糖酶(Xylanase,xynB)[19,20]作为外源蛋白的模型,对PAOX2上游调控序列进行随机突变,构建了PAOX2突变型毕赤酵母文库,根据表达木聚糖酶酶活的差别,筛选突变菌株并对其PAOX2进行测序,最后分析突变位置对PAOX2表达能力的影响。
1 材料与方法
1.1 试剂和仪器
本研究实验的生化试剂盒、主要仪器见表1。
表1 试剂和仪器
1.2 载体及菌株
质粒pPIC9K、pPIC9K-PAOX2-xynB由实验室保藏;大肠杆菌Top 10购自TaKaRa公司;毕赤酵母 GS115菌株由实验室保藏。
1.3 培养基与溶液的配制
Luria-Bertani(LB)培养基:称取1 g蛋白胨、0.5 g酵母粉、1 g氯化钠,溶于100 mL去离子水水中,调整pH至7.0。如制作平板,则加入1.5 g琼脂粉。121 ℃高压灭菌15 min。
Yeast Extract Peptone Dextrose (YPD)培养基:称取1 g 酵母粉、2 g蛋白胨、2 g葡萄糖,溶于100 mL去离子水中。如需固体培养基,则加入1.5 g琼脂粉。115 ℃高压灭菌20 min。配置好的YPD培养基于室温存放,平板置于4 ℃储存。
Minimal Dextrose(MD)培养基:称取1.34 g YNB、2 g葡萄糖、1.5 g琼脂粉(配制平板时添加)溶解于100 mL去离子水中,115 ℃高压灭菌20 min。使用前加入200 μL浓度为4×10-5g/L的无菌生物素。配置好的培养基于室温存放,平板置于4 ℃冰箱存放。
磷酸盐缓冲液 (PBS):称取8 g氯化钠、0.2 g氯化钾、3.63 g十二水合磷酸氢二钠、0.24 g磷酸二氢钾,溶解于900 mL去离子水中,用盐酸调整pH,室温存放。
Buffered Glycerol-complex (BMGY)培养基:称取酵母粉1 g、蛋白胨2 g溶解于70 mL 去离子水中,115 ℃高压灭菌20 min。量取10 mL 1 mol/L PBS缓冲液(pH视情况调整),115 ℃高压灭菌20 min。称取1.34 g YNB溶解于10 mL去离子水中,115 ℃高压灭菌20 min。称取1.26 g甘油溶解于9 mL 去离子水中,115 ℃高压灭菌20 min。上述四种灭菌试剂于室温存放,使用前将其混合到一起并加入200 μL无菌的 4×10-5g/L 生物素,充分混匀。
柠檬酸-磷酸缓冲溶液:称取2.9244 g磷酸氢二钠、2.04 g柠檬酸溶解于200 mL去离子水中,充分溶解混匀后则制成pH为5.0,浓度为50 mmol/L的柠檬酸-磷酸缓冲溶液,室温存放。
木聚糖溶液:将1 g木聚糖溶解于100 mL pH为5.0,浓度为50 mmol/L的柠檬酸-磷酸缓冲溶液中,充分混匀制成1% 的木聚糖溶液。由于木聚糖溶液稳定性不高,应尽量在使用前配制,室温存放。
木糖溶液:准确称量分析纯的无水木糖100 mg(预先在105 ℃干燥至恒重),加入少量去离子水溶解,定容至100 mL,充分摇匀后即获得浓度为1 mg/mL的木糖溶液。
DNS:将1.071 g/mL的苯酚于50 ℃充分融化,称取91.1 g酒石酸钾钠、3.15 g 3,5-二硝基水杨酸溶解于去离子水中,加入10.5 g氢氧化钠和2.33 mL融化的苯酚,再加入2.5 g偏重亚硫酸钠,将溶液定容至500 mL。配制好的溶液应存放于4 ℃,并于配好后7 d使用,有效期为6个月。
1.4 PAOX2序列的随机突变
以重组质粒pPIC9K-PAOX2-xynB为模板,TB-AOX2-F/R (TB-AOX2-F:5′-tcgagatctgagctcgaattctttt-3′;TB-AOX2-R:5′-tctcatcgtttggatccttttctcagt-3′)为引物,利用Diversify®随机突变试剂对PAOX2进行PCR。PCR反应程序为94 ℃预变性30 s;94 ℃变性30 s,54 ℃退火延伸96 s,共循环25次;最后再54℃延伸100 s,4℃冷却。将扩增后的产物经过2%琼脂糖凝胶电泳分析,并利用PCR清洁试剂盒对随机扩增产物进行纯化。
重组质粒pPIC9K-PAOX2-xynB经限制性内切酶BamH 1和EcoR 1消化后,回收双酶切位点BamH 1之间的片段(593 bp)以及酶切位点BamH 1和EcoR 1之间的片段(8 295 bp),然后利用T4连接酶对以上两个片段进行连接(16 ℃连接4 h)。利用PCR清洁试剂盒对T4酶连接后的产物进行纯化,然后利用In-fusion技术对纯化后的连接产物与突变后的PAOX2片段进行连接。然后将此连接产物转化至大肠杆菌TOP 10感受态中,并在含有氨苄的抗性平板上筛选。将平板置于37 ℃培养箱中12 h~16 h,待平板上长出单菌落之后挑取单菌落,以TB-AOX2-F/R为引物进行菌落PCR验证并测序,测序由生工生物工程(上海)股份有限公司完成。
1.5 PAOX2突变型毕赤酵母的构建和培养
PAOX2突变后的重组质粒经Sal1酶切线性化后,电转至毕赤酵母 GS115感受态细胞后涂MD平板培养基。MD平板培养基经30℃培养至长出单菌落,分别挑取突变型毕赤酵母单菌落接种于50 mL YPD培养基中,在30 ℃、200 r/min摇床中培养24 h。将突变型毕赤酵母种子液按4%的接种量接种到20 mL BMGY培养基中,在30 ℃、200 r/min摇床中培养48 h,使其培养基中的甘油耗尽,此时添加1%的甲醇进行诱导,之后每隔24 h补加1%甲醇,共诱导96 h。在每次补加甲醇之前将发酵液充分吸打混匀,然后取样。最后于4 ℃,12 000 g条件下离心发酵液10 min。收集上清,测定样品中的木聚糖酶酶活。每株菌株的诱导设计三个平行样。
1.6 木聚糖酶酶活力定义及测定方法
向2 mL 1% 的木聚糖溶液中加入20 μL 发酵液离心上清(空白对照组用去离子水代替),迅速吸打混匀后,50 ℃水浴5 min。迅速加入2 mL DNS溶液和1 mL 水后吸打混匀,100 ℃水浴 10 min 显色。然后迅速取出加入去离子水定容至20 mL。充分混匀后,利用酶标仪在540 nm处测定光吸收值,最后计算每个样品的酶活。每个反应测定三个平行样。木聚糖酶酶活力单位的定义为:在标准条件下,每分钟水解1%木聚糖形成1 μmol/L木糖(还原糖)所需酶量为1个酶活力单位[21]。
2 结果与分析
2.1 PAOX2的随机突变及PAOX2突变型毕赤酵母的构建
通过改变PCR反应中Mn2+和dGTP的量来控制随机突变水平,对PAOX2进行随机突变。扩增后的产物经2% 琼脂糖凝胶电泳分析显示突变后的PAOX2大小约为1 550 bp(图1-a)。重组质粒pPIC9K-PAOX2-xynB被BamH 1酶和EcoR 1消化后,经1% 琼脂糖凝胶电泳分析显示酶切产物共有三个片段,大小分别约为 8 300 bp,1 500 bp和600 bp(图1-b)。将双酶切位点BamH 1之间的片段(593 bp)以及酶切位点BamH 1和EcoR 1之间的片段(8 295 bp)进行T4连接,连接产物经1%琼脂糖凝胶电泳分析显示其大小约为8 900 bp(图1-c)。T4连接产物与突变后的PAOX2片段经In-fusion技术连接后转化大肠杆菌,随机挑取9株大肠杆菌进行菌落PCR验证,验证结果如图1-d所示,PCR产物经2%琼脂糖凝胶电泳分析显示其大小约为1 600 bp。测序显示其中4株菌株的PAOX2已经发生突变,表示约有50%的片段已发生突变,说明成功构建了pPIC9K-PAOX2-xynB启动子突变文库。
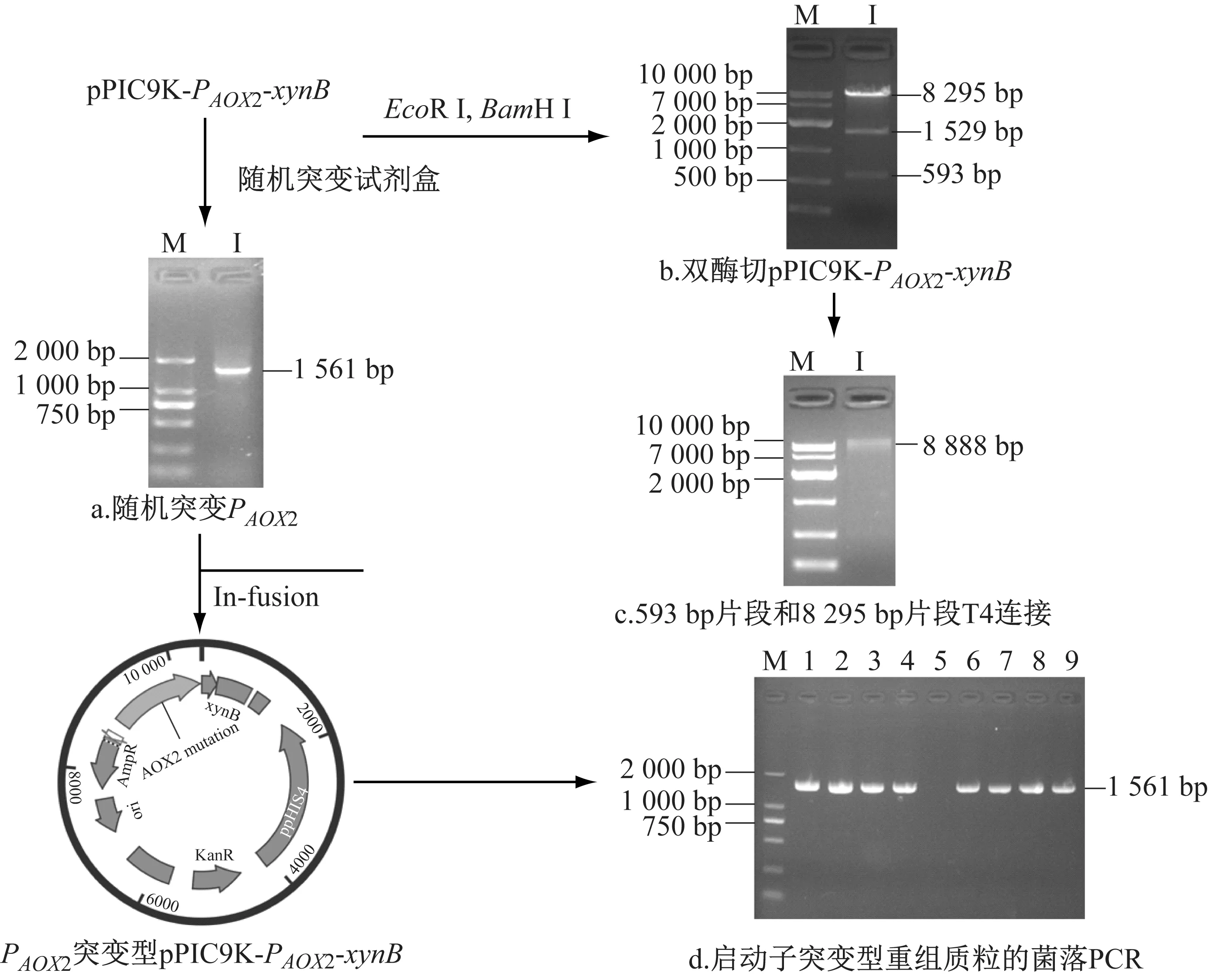
图1 PAOX2突变质粒的构建
将启动子突变型重组质粒酶切后电转入毕赤酵母中,即获得了PAOX2突变型毕赤酵母文库(图2)。
图2 PAOX2突变型毕赤酵母
2.2 PAOX2突变型毕赤酵母的筛选
以野生型PAOX2为启动子的重组毕赤酵母GS115-pPIC9K-PAOX2-xynB为对照组,挑取了850株PAOX2启动子突变型毕赤酵母进行了培养。图3展示了850株PAOX2突变型毕赤酵母表达木聚糖酶的活力,按照活力的高低分为6组。对照组酶活的0%~20%、20%~40%、40%~60%、60%~80%、80%~100%、100%~120%区间分别有207、58、43、44、260、238株突变型酵母菌。约有59%的突变菌株表达的木聚糖酶活力为对照组菌株的80%~120%,约有24%的突变菌株表达的木聚糖酶活力为对照组菌株的0%~20%,最后,约有17%的突变型菌株表达的木聚糖酶活力为对照组菌株的20%~80%,此区间内的酶活显著低于或稍微低于对照组菌株的酶活,这可能是由于突变造成了PAOX2表达木聚糖酶的能力下降。图4展示了20%~40%、40%~60%和60%~80%三个组别的菌浓度,他们的OD600分别为29.45±2.24、30.02±4.22和28.79±3.44,菌浓度基本相同,说明此次PAOX2随机突变对菌的生长没有明显影响。
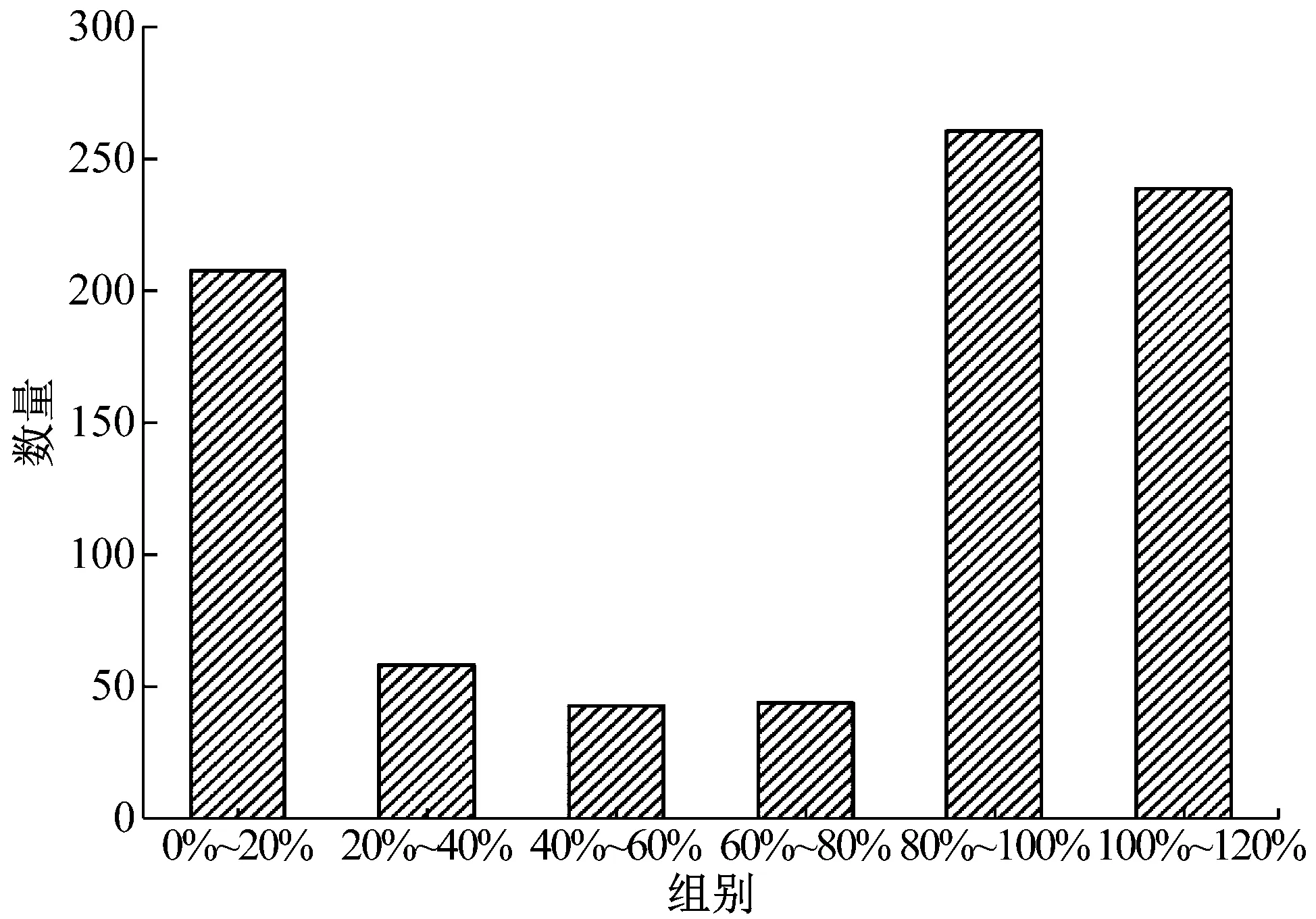
图3 不同表达量的菌株数量
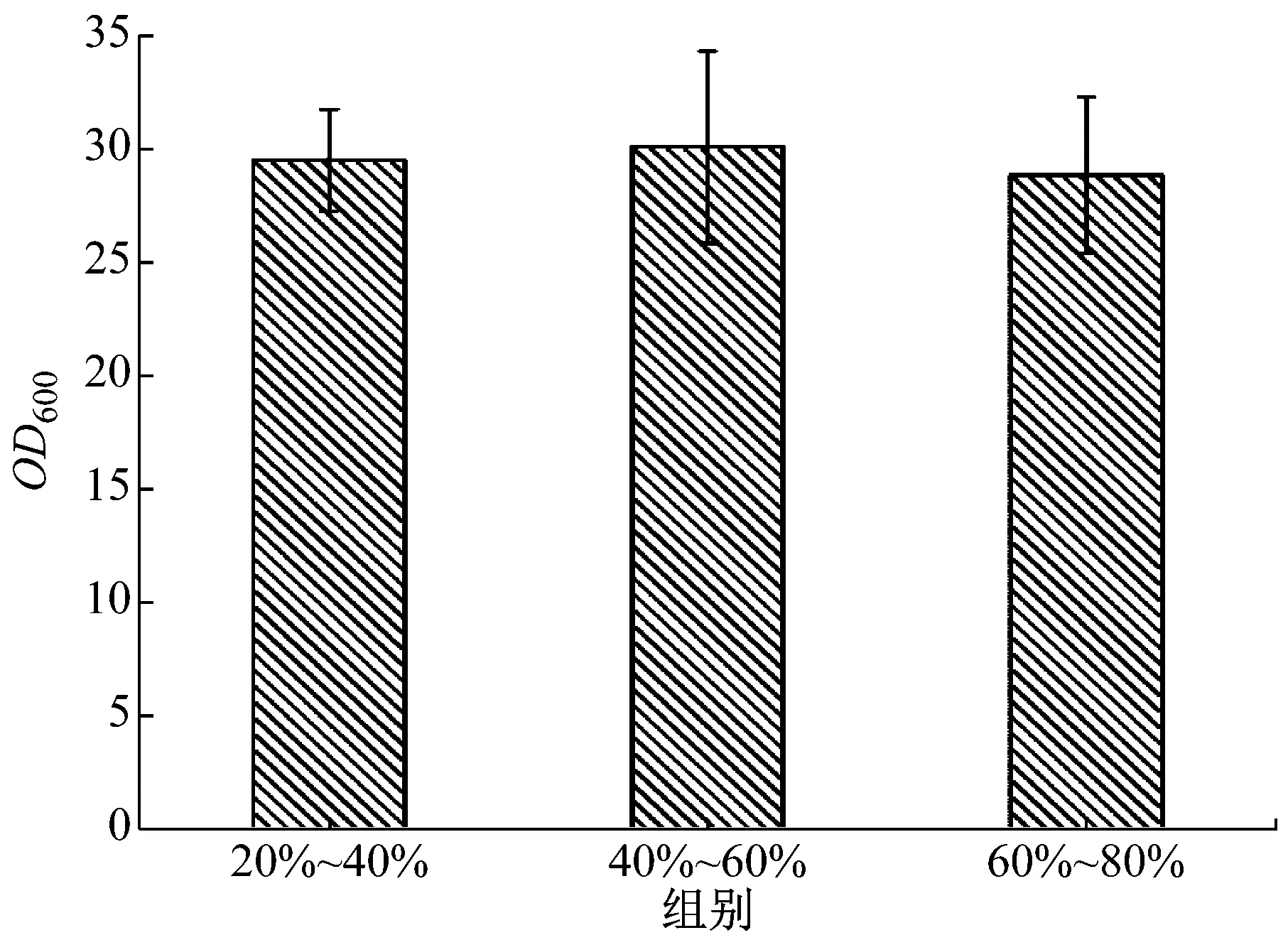
图4 20%~40%、40%~60%和60%~80%组别菌的浓度
2.3 毕赤酵母PAOX2上游调控序列分析
测序结果显示共有两株菌株(20-16和20-20)的PAOX2发生了突变,其与NCBI公布(模板质粒的PAOX2已测序,显示其与NCBI公布的PAOX2完全一致)的PAOX2序列比对结果(如图5所示)。PAOX2共有1 550个碱基,突变型菌株20-16在702(即-849)位置发生碱基T的插入导致其表达木聚糖酶活力为对照组菌株的36%突变型菌株20-20在789(即-762)位置插入了碱基A,其表达木聚糖酶的酶活为对照组菌株的10%。OHI等[15]鉴定出一个上游激活元件(UAS)和两个上游抑制元件(URS)。UAS位于PAOX2的-341至-273之间,其中,处于-337至-313位置的序列GATAGGCTATTTTTGTCGCATAAAT是PAOX2UAS的重要组成部分,且处于PAOX2的-293至-283之间的序列不参与转录调控,PAOX2不会因为此片段的缺失而提高表达能力。两个URS分别位于PAOX2的-780至-342之间和-255至-215之间。本研究的结果表明在-849~-762之间的碱基序列也对PAOX2的转录有重要作用。

图5 毕赤酵母PAOX2与测序菌株的PAOX2的比对结果
本研究中仍有大量未发生突变的PAOX2的片段导致木聚糖酶活力显著下降,这可能由于大批量培养重组毕赤酵母可能会导致平行性出现较大差异,重组毕赤酵母表达木聚糖酶活力产生波动。
3 结论
通过易错PCR的方式对PAOX2上游调控序列进行随机突变,构建了PAOX2突变文库,筛选了850株重组菌。测序结果表明突变型菌株在-847插入碱基A或者-788插入碱基T分别使木聚糖酶活力下降为10%和36%。这说明-847 ~-788片段对PAOX2的转录有重要作用。
猜你喜欢
昆明医科大学学报(2020年12期)2021-01-26
广西医学(2020年13期)2020-03-04
中国病理生理杂志(2018年9期)2018-09-27
食品与发酵工业(2018年3期)2018-04-12
上海农业学报(2017年4期)2017-04-10
广东饲料(2016年1期)2016-12-01
工业微生物(2016年5期)2016-11-11
化学与生物工程(2016年10期)2016-11-10
中国粮油学报(2016年5期)2016-01-23
中国现代医生(2015年5期)2015-03-31
