
高效液相色谱法测定食用油中抗氧化剂的国家标准方法的优化研究
2021-06-29 10:16钟慈平成长玉周海燕余晓琴
理化检验-化学分册 2021年6期
钟慈平,成长玉,苏 欣,周海燕,马 伟,余晓琴∗
(1.四川省食品检验研究院,成都 611731;2.四川农业大学 食品学院,雅安 625014)
油脂受氧气、光、热、微生物等影响会逐渐水解或氧化而变质酸败,进而分解为甘油和油脂酸,然后油脂酸中不饱和链断开形成过氧化物,再依次分解为低级油脂酸、醛类、酮类等物质[1-2],从而产生异味、改变感官性质、破坏营养物质,甚至可能致癌[3]。抗氧化剂可分为天然抗氧化剂(维生素E、茶多酚等)和合成抗氧化剂[4-5],其作为食品添加剂,能防止油脂自动氧化并有效增加脂类物质的氧化稳定性。因合成抗氧化剂更易制得、价格低廉、抗氧化能力更强而被广泛使用,同时为了达到更好的抗氧化效果,生产者会在食品中添加多种抗氧化剂复配使用[6]。动物试验证明,使用过量抗氧化剂会诱发癌症等疾病,对人体有较大潜在的危害[7-9]。我国食品添加剂标准GB 2760-2014 规定叔丁基对苯二酚(TBHQ)、叔丁基对羟基茴香醚(BHA)和2,6-二叔丁基对甲基苯酚(BHT)在油脂中的残留量不超过0.2 g·kg-1,没食子酸丙酯(PG)在油脂中的残留量不超过0.1 g·kg-1[10]。食用油是油脂的大众消费主体,因此很有必要建立一套准确、快速、稳定测定食用油中抗氧化剂含量的方法。
目前,检测抗氧化剂的方法较多,包括气相色谱法[11]、气相色谱-串联质谱法[12]、高效液相色谱法[13-14]、高效液相色谱-串联质谱法[15]、电化学法[16]和比色法[17]等。比色法灵敏度低、准确度差,不能同时测定多种抗氧化剂;电化学法灵敏度较高,但选择性差,电解液毒性较大,对环境污染大。国家标准GB 5009.32-2016《食品安全国家标准 食品中9种抗氧化剂的测定》[18]自2017年实施以来,检测机构在使用过程中发现,此标准第一法(高效液相色谱法)回收率普遍偏低,部分样品存在明显的干扰(具体见2.1 节)。针对上述问题,本工作对GB 5009.32-2016第一法主要做了以下优化:①以含L-抗坏血酸棕榈酸酯(AP)的正己烷饱和的乙腈溶液(配制方法见1.1节)作为提取剂,AP能降低抗氧化剂在提取和净化浓缩过程中的损失;②增加提取剂用量;③合并收集净化步骤过程中的淋洗液和洗脱液,大大提高回收率;④筛选合适色谱柱和优化流动相,改善分离度。
通过以上改进,食用油中2,4,5-三羟基苯丁酮(THBP)、去甲二氢愈创木酸(NDGA)、PG、TBHQ、没食子酸十二酯(DG)、没食子酸辛酯(OG)、2,6-二叔丁基-4-羟甲基苯酚(Ionox-100)、BHA 和BHT 等常见的9种抗氧化剂回收率提高至62.0%~104%,分离度满足要求,其结果令人满意。
1 试验部分
1.1 仪器与试剂
Agilent 1260 型高效液相色谱仪;MULTIFUGEX3R 型离心机;T25 Digital型涡旋振荡仪;N1型氮吹仪;ML-204型电子天平(感量0.001 g);C18固相萃取柱(500 mg/6 mL)。
抗氧化剂标准储备溶液:2 000 mg·L-1,称取BHA、BHT、OG、DG、PG、NDGA、THBP、TBHQ和Ionox-100等9种抗氧化剂标准品各20.00 mg,分别置于10 mL 棕色容量瓶中,用含0.1 g·L-1AP 的乙腈溶液定容,配制成质量浓度为2 000 mg·L-1的抗氧化剂标准储备溶液,于-18 ℃避光保存。
抗氧化剂混合标准溶液:200 mg·L-1,分别移取上述9种抗氧化剂标准储备溶液各1 mL 置于同一10 mL容量瓶中,用含0.1 g·L-1AP的乙腈溶液定容,摇匀,配制成质量浓度为200 mg·L-1的抗氧化剂混合标准溶液,现配现用。
抗氧化剂混合标准溶液系列:将200 mg·L-1抗氧化剂混合标准溶液用含0.1 g·L-1AP的乙腈溶液逐级稀释,配制成质量浓度分别为2.00,5.00,10.0,20.0,40.0,80.0 mg·L-1的抗氧化剂混合标准溶液系列,现配现用。
乙腈饱和的正己烷溶液:取800 mL正己烷,加入200 mL乙腈,摇匀静止后取上层即得。
正己烷饱和的乙腈溶液:取800 mL乙腈,加入200 mL正己烷,摇匀静止后取下层即得。
食用油购自某市场。
BHA、BHT、OG、DG、PG、NDGA、THBP、TBHQ 和Ionox-100标准品的纯度均不小于98%;AP为分析纯;甲醇、正己烷和乙腈为色谱纯。
1.2 仪器工作条件
Agilent Eclipse XDB-C18色 谱柱(250 mm×4.6 mm,5μm);柱温35 ℃;进样量10μL;检测波长280 nm;流量1 mL·min-1;流动相A 为0.1%(体积分数,下同)甲酸溶液,B为乙腈。梯度洗脱程序:0~4.00 min时,B为5%;4.00~12.00 min时,B由5%升至60%;12.00~22.00 min时,B由60%升至75%;22.00~25.00 min时,B由75%升至95%;25.00~28.10 min 时,B 由95%降 至5%,保 持1.90 min。
1.3 试验方法
1.3.1 样品提取
称取试样1.000 g置于50 mL 离心管中,加入5 mL乙腈饱和的正己烷溶液溶解样品,涡旋1 min,用10 mL正己烷饱和的乙腈溶液(含0.1 g·L-1AP)涡旋提取2 min,以转速6 000 r·min-1离心5 min,收集乙腈层置于试管中,再重复使用10 mL正己烷饱和的乙腈溶液(含0.1 g·L-1AP)提取2次,合并3 次提取液,待净化。同时做空白试验。
1.3.2 样品净化
用甲醇5 mL 活化C18固相萃取柱,再用乙腈5 mL平衡,弃去流出液。将上述合并的提取液倾入C18固相萃取柱中,用体积比2∶1的乙腈-甲醇混合液5 mL洗脱,收集上样时的流出液和洗脱液置于试管中,于40℃氮吹至近干,用含0.1 g·L-1AP的乙腈溶液2 mL定容,经0.22μm 有机滤膜过滤,按仪器工作条件进行测定。
2 结果与讨论
2.1 GB 5009.32-2016第一法的试验结果
参照GB 5009.32-2016第一法的样品提取净化过程和高效液相色谱条件进行空白基质(阴性食用油)加标试验,考察了该方法下9种抗氧化剂的分离效果(图1),并计算得到9种抗氧化剂的加标回收率。

图1 采用GB 5009.32-2016第一法时9种抗氧化剂的色谱图Fig.1 Chromatogram of 9 antioxidants by the first method of GB 5009.32-2016
结果显示:在完全采用GB 5009.32-2016第一法的色谱柱和流动相洗脱程序情况下,PG 峰形较差,THBP峰和TBHQ 峰未能达到基线分离,DG和BHT 出峰较晚;另外,9种抗氧化剂的加标回收率偏低(31.4%~90.1%),尤其是TBHQ、Ionox-100和BHT 的加标回收率均低于70.0%。
2.2 色谱条件的选择
2.2.1 色谱柱
试验首先选用较短的Agilent Eclipse XDB-C18色谱柱(150 mm×4.6 mm,5μm)对抗氧化剂混合标准溶液和实际食用油样品进行分析,所得色谱图见图2。
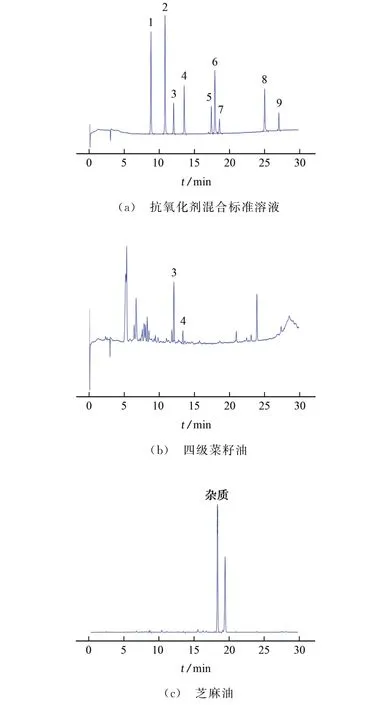
图2 采用柱长为150 mm 的色谱柱时的色谱图Fig.2 Chromatograms obtained by chromatographic column with the column length of 150 mm
由图2可知:混合标准溶液中的9种抗氧化剂能得到较好分离,但实际样品中食用油基质组分复杂,出现明显干扰,如四级菜籽油中TBHQ 峰与基质组分未达到完全分离;芝麻油中基质组分在Ionox-100出峰处(18.554 min)出现一未知杂质(经质谱验证不是Ionox-100),严重影响Ionox-100 的测定。
为获得良好分离,试验考察了Agilent Eclipse XDB-C18、SHISEIDO C18、Waters C18等3种柱长为250 mm 的色谱柱对9 种抗氧化剂分离效果的影响。结果表明:采用SHISEIDO C18和Waters C18色谱柱测定时,部分抗氧化剂(如PG、THBP)的峰形较差,THBP峰和TBHQ 峰不能完全分开,影响积分结果,定量分析误差大;而采用Agilent Eclipse XDB-C18柱测定时,9种抗氧化剂全部出峰,出峰时间合适,峰形对称,分离度相对较好。因此,试验选用Agilent Eclipse XDB-C18柱(250 mm×4.6 mm,5μm)作为色谱柱。
2.2.2 流动相
试验考察了1.5%(体积分数,下同)乙酸-甲醇、1.5%乙酸-甲醇与乙腈混合液(体积比为7∶3)、0.1%甲酸-乙腈等3种流动相体系对9种抗氧化剂分离效果的影响,所得色谱图见图3。
由图3可知:在其他色谱条件相同的情况下,以1.5%乙酸-甲醇体系、1.5%乙酸-甲醇与乙腈混合液(体积比为7∶3)体系为流动相时,9种抗氧化剂出峰时间延迟,且THBP峰与TBHQ 峰部分重叠;以0.1%甲酸-乙腈体系为流动相时,9种抗氧化剂能清晰完全分离,并且峰形对称。因此,试验选择0.1%甲酸溶液-乙腈体系作为流动相,在30 min内完成9种抗氧化剂的测定。

图3 3种流动相下9种抗氧化剂的色谱图Fig.3 Chromatograms of 9 antioxidants with 3 mobile phases
2.2.3 柱 温
试验考察了柱温分别为30,35 ℃时对80.0 mg·L-1抗氧化剂混合标准溶液的分离效果的影响。结果表明:当柱温为30 ℃时,出峰时间接近的BHA 峰和OG 峰不能完全分开,影响定量分析;当柱温为35℃时,各组分能较好分离,且峰形较好,没有明显的拖尾现象。因此,试验选择柱温为35 ℃。
2.2.4 洗脱梯度
由于测定芝麻油和四级菜籽油时存在较为明显的基质干扰,杂质与部分抗氧化剂的保留时间接近(四级菜籽油中TBHQ、芝麻油中Ionox-100出峰处的杂峰干扰),影响定量,因此对梯度洗脱程序进行优化。结果表明:按照1.2节的梯度洗脱程序测定含基质干扰的抗氧化剂混合标准溶液,各组分均能较好分离,满足准确定性和定量的检测要求。
2.3 提取条件的选择
试验选用含AP的正己烷饱和的乙腈溶液为提取剂,与GB 5009.32-2016中第一法的提取剂正己烷饱和的乙腈溶液进行比较,其回收试验结果见图4。
由图4可知:经正己烷饱和的乙腈溶液提取后,THBP、THBQ、DG 和BHT 的回收率较低;而经含AP的正己烷饱和的乙腈溶液提取后,9种抗氧化剂的回收率均高于85.0%,满足检测要求,原因是AP能保护抗氧化剂在提取浓缩等过程中不被氧化。因此,试验选用含AP 的正己烷饱和的乙腈溶液作为提取剂。

图4 不同提取剂下9种抗氧化剂回收率的比较Fig.4 Comparison of recoveries of 9 antioxidants with different extractants
GB 5009.32-2016第一法中提取剂的用量为5 mL,试验进一步考察了提取剂含AP的正己烷饱和的乙腈溶液的用量分别为5,10 mL 时对回收试验结果的影响。结果表明:当使用10 mL 提取剂时,9种抗氧化剂的回收率均有提高,其中BHT 的回收率显著上升。因此,试验选择提取剂的用量为10 mL。
2.4 净化柱的选择
试验考察了C18、C18-N、PSD、改性C18等不同净化柱对回收试验结果的影响,见图5。
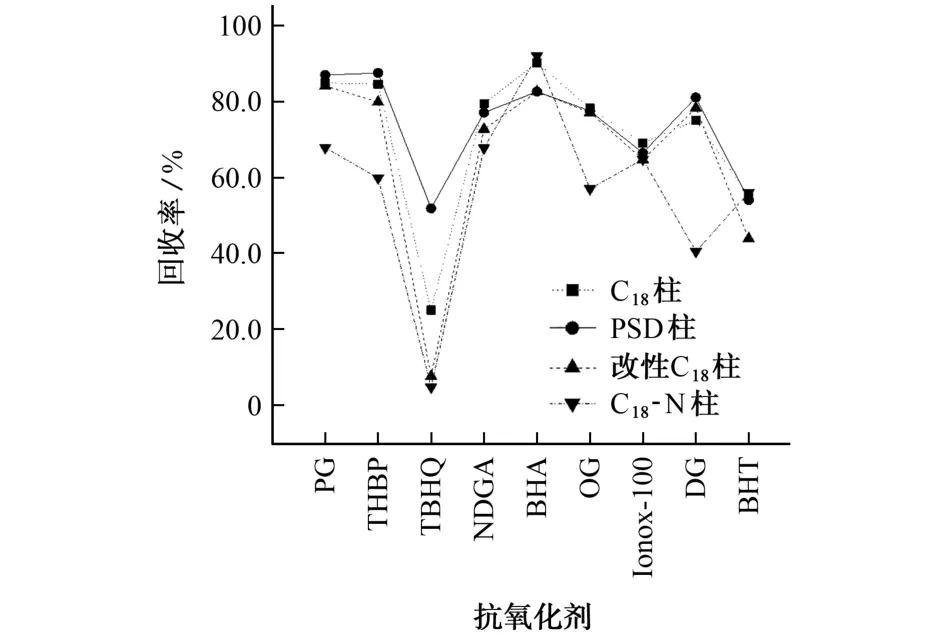
图5 采用不同净化柱时9种抗氧化剂回收率的比较Fig.5 Comparison of recoveries of 9 antioxidants with different purified columns
由图5可知:使用C18柱和PSD 柱均能得到较高的回收率,C18主要吸附长链脂类化合物、甾醇及其他非极性杂质,PSD 主要吸附脂肪酸、脂类、有机酸、色素等物质。另外,由于C18柱在GB 5009.32-2016中使用,因此试验选择C18柱作为净化柱。
2.5 氮吹浓缩过程的影响
在40 ℃氮吹下,按试验方法分别测定未浓缩、添加AP浓缩和未添加AP浓缩的10.0 mg·L-1抗氧化剂混合标准溶液中9种抗氧化剂的含量。结果显示,9种抗氧化剂在上述情况下测得的峰面积变化不大,说明40℃氮吹浓缩过程对9种抗氧化剂并未带来明显损失。
2.6 标准曲线和检出限
按试验方法对抗氧化剂混合标准溶液系列进行测定,以抗氧化剂的质量浓度为横坐标,对应的峰面积为纵坐标绘制标准曲线。结果表明:9种抗氧化剂的质量浓度在2.00~80.0 mg·L-1内与峰面积呈线性关系,相关系数均在0.999 0以上,具体的线性参数见表1。
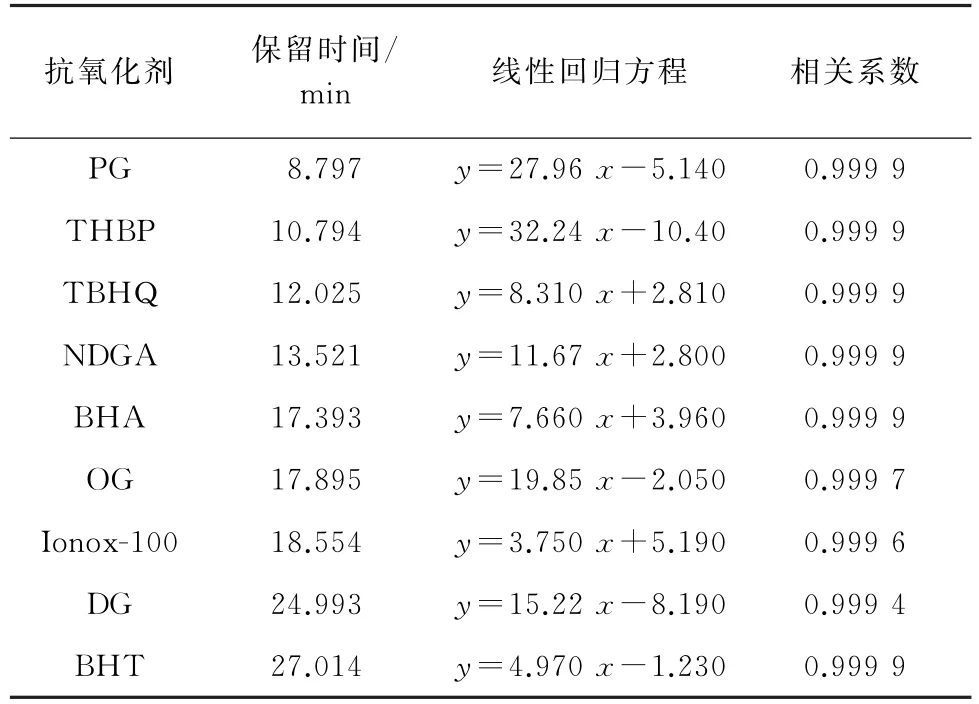
表1 保留时间和线性参数Tab.1 Retention times and linearity parameters
以3倍信噪比(S/N)对应的质量浓度作为检出限(3S/N),得到9 种抗氧化剂的检出限均为10.0 mg·kg-1。
2.7 稳定性试验
在一段时间内(0,10,20,30,40,50,60 h),分别对样品溶液和抗氧化剂混合标准溶液(质量浓度10.0 mg·L-1,含AP的乙腈配制)的稳定性进行考察。结果显示,随着时间的延长,样品溶液和抗氧化剂混合标准溶液的质量浓度无显著变化,不同时间点测定值的相对标准偏差(RSD,n=6)均小于10%,说明60 h内9种抗氧化剂的稳定性良好。
2.8 精密度和回收试验
在芝麻油、玉米油、一级菜籽油、四级菜籽油、胡麻油、橄榄油、植物调和油等7种食用油空白基质中分别添加20.0,60.0,150.0 mg·kg-1等3个浓度水平的抗氧化剂混合标准溶液,按试验方法测定,其回收率和测定值的RSD 结果见表2。
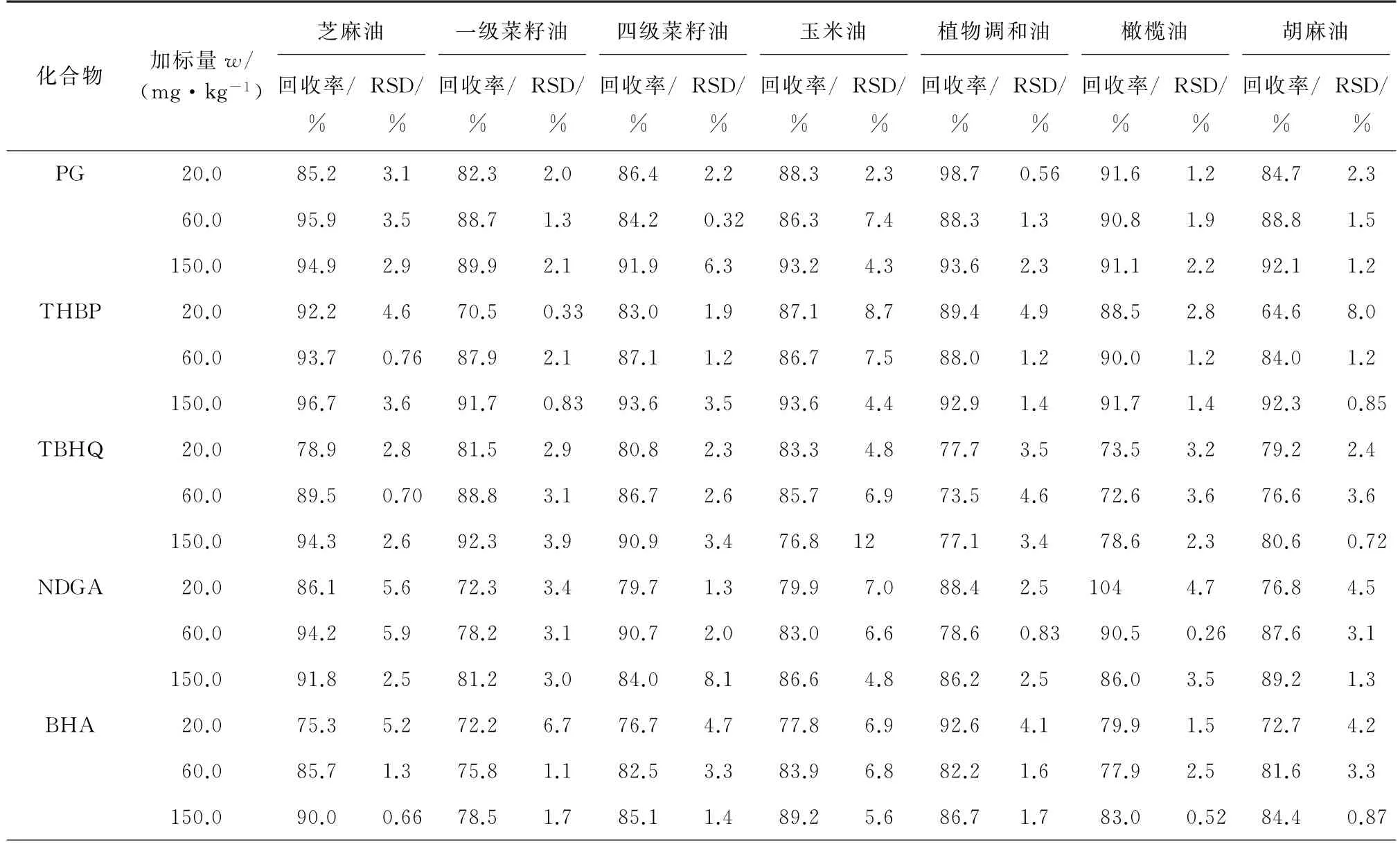
表2 精密度和回收试验结果(n=6)Tab.2 Results of tests for precision and recovery(n=6)

表2 (续)
由表2 可知:9 种抗氧化剂的回收率为62.0%~104%,RSD 均小于13%,满足测定要求。
2.9 样品分析
采用优化后的试验方法对市售不同种类、不同级别和不同厂家的食用油中抗氧化剂含量进行测定,包括一级菜籽油、四级菜籽油、胡麻油、橄榄油、植物调和油、玉米油和芝麻油各2批。结果表明:大部分样品中未检出PG、THBP、NDGA、BHA、OG、Ionox-100、TBHQ、BHT,仅2批芝麻油中检出DG,检出量分别为17,19 mg·kg-1。
本工作优化了GB 5009.32-2016第一法中的色谱条件、提取剂、净化浓缩步骤,考察了优化后方法的稳定性、加标回收率、线性范围、精密度等。结果表明该方法操作简单、准确度高、分离效果好、灵敏度高,满足检测标准要求,适用于同时检测大批量食用油中多种抗氧化剂的含量。
猜你喜欢
石油炼制与化工(2022年2期)2022-02-15
应用化工(2021年4期)2021-05-20
化工管理(2020年26期)2020-10-09
石油沥青(2020年1期)2020-05-25
伴侣(2019年10期)2019-10-16
今日农业(2019年12期)2019-08-15
中国粮油学报(2019年4期)2019-07-12
山东化工(2019年2期)2019-02-21
今日农业(2019年14期)2019-01-04
中国药理学与毒理学杂志(2015年3期)2015-12-16
