
结核分枝杆菌感染对痰液菌群结构的影响
2021-04-07 10:08张晨晨谭卫国郭卉欣黄新春陈燕梅魏文静
中国防痨杂志 2021年4期
张晨晨 谭卫国 郭卉欣 黄新春 陈燕梅 魏文静
近年许多研究显示,除受结核分枝杆菌(Mycobacteriumtuberculosis,MTB)感染影响外,结核病也可能是由复杂的微生物群落相互作用导致的[1]。早期肺结核患者呼吸道菌群的相关研究多是基于分离培养方法。但是到目前为止,人类很难模拟各种微生物的原始生存环境,必然也不能真实地还原肺结核患者呼吸道本身微生物群系特点。分子生物学技术的发展和应用,极大的提升了对人呼吸道菌群的研究水平,如在新生儿感染性肺炎、婴幼儿喘息、呼吸机相关性肺炎、细菌性重症肺炎和哮喘的研究方面都取得了一定的成果[2-6]。笔者利用16S rRNA高通量测序技术,比较了健康人群、结核分枝杆菌潜伏感染(LTBI)者和活动性肺结核患者的痰液菌群变化,初步研究了MTB感染对人痰液菌群结构的影响,以为结核病防治工作提供参考。
对象和方法
1.研究对象:搜集2016年11月至2017年12月深圳市慢性病防治中心健康体检者53名作为A组,其中,男14名(26.4%),女39名(73.6%),年龄中位数(四分位数)为39.0(30.0,49.0)岁;选取同期该中心登记确诊的结核菌素皮肤试验(tuberculin skin test,TST)硬结平均直径≥10 mm和γ干扰素释放试验(interferon gamma release assay,IGRA)均为阳性的LTBI者41例作为B组,其中,男18例(43.9%),女23例(56.1%),年龄中位数(四分位数)为45.0(31.5,54.0)岁;初治活动性肺结核患者54例作为C组,其中,男36例(66.7%),女18例(33.3%),年龄中位数(四分位数)为31.5(24.8,42.3)岁。
2.纳入标准:WHO在2018年更新的《潜伏性结核感染管理指南》[7]中明确给出了LTBI检测和治疗人群的界定,活动性肺结核的诊断参照《WS 288—2017 肺结核诊断》[8]。所有研究对象均符合以下纳入标准:深圳市本地人口,作息、饮食规律,不酗酒嗜烟;无心、肝、肺、肾功能不全等重大疾病;无糖尿病、恶性肿瘤、HIV感染等。入组前所有研究对象均被告知研究内容后同意参加,并签署知情同意书。本研究符合《涉及人的生物医学研究伦理审查办法》和《世界医学协会赫尔辛基宣言》的相关规定。
3.标本采集:告知研究对象清晨空腹,用生理盐水漱口后采用用力深咳方式,收集其下呼吸道痰液样本(以干酪痰和黏液痰为佳)3~5 ml,共148份,立即放入-80 ℃冰箱保存,供后期集中提取细菌基因组DNA,用于测序分析。
4.基因组DNA提取和PCR扩增:使用粪便DNA微型试剂盒(批号:116570200;美国MP Biomedicals 公司)提取痰液样品DNA。用正向引物515F(5′-GTGCCAGCMGCCGCGGTAA-3′)和反向引物907R(5′-CCGTCAATTCMTTTRAGTTT-3′)对细菌16S rRNA的V4~V5可变区进行PCR扩增,将样品特异性的7 bp条形码纳入引物中进行多重测序。PCR扩增反应体系50 μl,包括:PCR 缓冲液 10 μl,dNTPs 10 μl,上下游引物各1.5 μl,Q5 High-Fidelity DNA 聚合酶 0.2 μl,模板DNA 40 ng,ddH2O补充至总体系50 μl。扩增程序为:98 ℃预变性2 min;98 ℃变性15 s,55 ℃退火30 s,72 ℃延伸30 s,25个循环;最后72 ℃延伸5 min。PCR扩增产物用核酸纯化试剂盒Agencourt AMPure Beads(批号:A63881;美国Beckman Coulter公司)纯化,使用PicoGreen dsDNA检测试剂盒(批号:P7581;美国Invitrogen公司)进行定量分析。
5.16S rRNA宏基因组测序和数据处理:取等量纯化后产物进行Illlumina MiSeq平台高通量测序(上海派森诺生物科技有限公司)。使用微生物生态学软件(QIIME 2 2019.10)对细菌16S rRNA扩增子数据进行生物信息学分析。使用FastQC v.0.11.2和Trimmomatic v.0.32对序列进行质量控制和过滤,然后使用QIIME 2内带的DADA2软件对序列reads进行过滤,构建特征表。采用q2-feature-classifier模块(QIIME 2 2019.10)对每个16S rRNA基因序列进行分类。采用预训练的朴素贝叶斯分类器gg-13-8-99-515-806-nb-classifier进行聚类分析,得到样本在不同分类水平上的菌群的相似性和差异性。
6.生物信息学分析:应用QIIME 2、Python(v3.7)和R软件(v3.6.3)进行序列数据分析。采用q2-diversity模块(QIIME 2 2019.10)进行Alpha和Beta多样性分析。Alpha多样性有多重衡量指标及测量方法,通过Shannon、Simpson、Chao1、Pielou_e和Observed_otus 5个指标来表示[9]。其中,Shannon指数是菌群丰富度和均衡度的量化指标,对丰富性更敏感,值越大,样本多样性越大;Simpson指数和Observed_otus是对菌群丰富度的定性测量,值越大,样本多样性越大;Chao1指数代表菌群丰富度指数,其值越大,表明物种总数越多;Pielou_e指数是菌群均匀度的衡量标准,值越高,菌群的均匀度越大。Beta多样性反映不同样品在物种多样性方面的相似程度[10],组间两两比较的Bray-Curtis距离差异采用非参数多元方差分析(PerMANOVA检验)。比较不同分类水平各物种的相对丰度,描述菌群组成结构的差异,然后通过线性判别分析效应量(linear discriminant analysis effect size,LEfSe)找出三组在各分类水平有明显差异的细菌,用线性判别分析(linear discriminant analysis,LDA)评估各组间各分类水平差异菌的效应大小,该研究设定差异有统计学意义的LDA 值为2,寻找两组间具有明显差异的物种[11-12]。
7.统计学处理:采用SPSS 23.0软件进行统计学分析,计量资料为偏态分布,以“中位数(四分位数)[M(Q1,Q3)]”表示,各组微生物多样性指数比较采用Kruskal-Wallis检验,以P<0.05为差异有统计学意义。
结 果
1. 16S rRNA 测序结果分析:通过高通量测序,对3个组的148份样本的原始数据进行处理,获得14 136 502.0条有效序列,A、B和C三组有效序列中位数(四分位数)分别为104 296.0(85 553.0,123 168.0)条、108 578.0(91 821.0,123 309.0)条和120 523.0(113 297.5,128 367.5)条。根据97%的相似度进行操作分类单元(operational taxonomic units,OTU)聚类,获得21 712.00个OTU,A、B和C三组OTU数的中位数(四分位数)分别为150.00(120.00,163.50)、137.00(114.50,167.50)和143.00(122.00,179.25)个,各组间OTU数比较,差异无统计学意义(H=0.440,P=0.803)。每份样本的测序覆盖率(coverage)均>99%,说明测序数目足够,测序序列可以代表其菌群组成。
2.菌群Alpha多样性分析:对以上测序数据,首先利用Alpha多样性分析法对不同组别间的菌群丰富度和均衡度进行分析,基于OTU种类和丰度计算Shannon、Simpson、Chao1、Pielou_e和Observed_otus指数。统计分析发现三组研究对象间呼吸道菌群多样性指数差异均无统计学意义(表1),组间两两比较发现A组和C组的Pielou_e指数间差异有统计学意义(H=4.462,P=0.035),说明健康人群和活动性肺结核患者呼吸道菌群的均匀度有明显差异。
3.菌群Beta多样性分析:Beta多样性反映了各样本间菌群的整体差异程度,图中两点之间的距离越大表示两者间的群落构成差异越大。基于Bray-Curtis距离指标分析(图1),可见A、B、C组研究对象整体菌群组成存在一定的差异(H=2.027,P=0.002)。
4.菌群结构组成:统计不同分类水平上各微生物的丰度(图2),结果显示,门水平上,三组样本中物种相对丰度较高的均为拟杆菌门(Bacteroidetes)、变形菌门(Proteobacteria)、厚壁菌门(Firmicutes)和梭杆菌门(Fusobacteria)。其中,B组厚壁菌门(Firmicutes)的相对丰度为20.4%,较A组(24.6%)和C组(25.3%)减少;与A组(10.2%)和B组(10.3%)相比,C组梭杆菌门(Fusobacteria)相对丰度(5.5%)明显减少。属水平上,相对丰度较高的属有20种,C组中卟啉单胞菌属(Porphyromonas)相对丰度为3.5%,纤毛菌属(Leptotrichia)相对丰度为1.7%,均比A组(8.0%和4.2%)和B组(8.0%和2.9%)减少;与A组(5.9%)和C组(3.7%)相比,B组梭杆菌属(Fusobacterium)相对丰度(7.3%)增加。在纲、目、科水平上,三组间不同物种的相对丰度也均有变化。
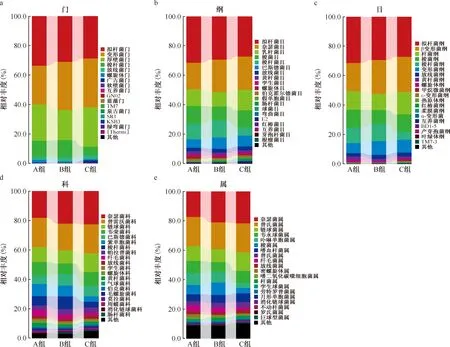
图2a表示门水平;图2b表示纲水平;图2c表示目水平;图2d表示科水平;图2e表示属水平;A组为健康组,B组为结核分枝杆菌潜伏感染组,C组为活动性肺结核组
5.不同分类水平上菌群组间差异物种分析:选取样本中相对丰度>1%的物种进行分析,用柱形图进行可视化展示(图3),在不同分类水平上筛选三组中丰富度变化差异较大的菌种,其中涉及3个门(厚壁菌门、梭杆菌门、螺旋体门)、3个纲(黄杆菌纲、梭杆菌纲、螺旋体纲)、5个目(黄杆菌目、乳杆菌目、梭杆菌目、伯克霍尔德菌目、螺旋体目)、6个科(紫单胞菌科、黄杆菌科、梭杆菌科、纤毛菌科、伯克菌科、螺旋体科)、5个属(二氧化碳噬纤维菌属、卟啉单胞菌属、梭杆菌属、纤毛菌属、密螺旋体属)和2个种(Nanceiensis和Parvula)。C组中梭杆菌门(Fusobacteria)[0.04(0.02,0.08)]、梭杆菌纲(Fusobacteriia)[0.04(0.02,0.08)]、梭杆菌目(Fusobacteriales)[0.04(0.02,0.08)]、梭杆菌科(Fusobacteriaceae)[0.02(0.01,0.05)]、紫单胞菌科(Porphyromonadaceae)[0.03(0.01,0.05)]、梭杆菌属(Fusobacterium)[0.02(0.01,0.05)]和卟啉单胞菌属(Porphyromonas)[0.03(0.01,0.05)]的丰富度较A组[丰富度分别为0.08(0.06,0.12)、0.08(0.06,0.12)、0.08(0.06,0.12)、0.04(0.03,0.06)、0.07(0.04,0.10)、0.05(0.03,0.06)、0.07(0.04,0.10)]和B组[丰富度分别为0.09(0.06,0.12)、0.09(0.06,0.12)、0.09(0.06,0.12)、0.05(0.03,0.08)、0.07(0.04,0.10)、0.05(0.03,0.09)、0.08(0.04,0.10)]明显降低,差异均有统计学意义(C组与A组比较:H值分别为14.780、14.780、14.732、11.489、24.910、11.321、25.726,P值均<0.01;C组与B组比较:H值分别为16.221、16.221、16.281、13.390、26.416、13.501、27.196,P值均<0.01)。而C组中伯克霍尔德菌目(Burkholderiales)[0.01(0.00,0.02)]和伯克菌科(Burkholderiaceae)[0.01(0.00,0.01)]的丰富度较A组[丰富度分别为0.00(0.00,0.01)和0.00(0.00,0.00)]和B组[丰富度分别为0.00(0.00,0.01)和0.00(0.00,0.00)]则明显增加,差异均有统计学意义(C组与A组比较:H值分别为9.733和4.799,P值分别为0.002和0.028;C组与B组比较:H值分别为12.134和8.152,P值均<0.01)。
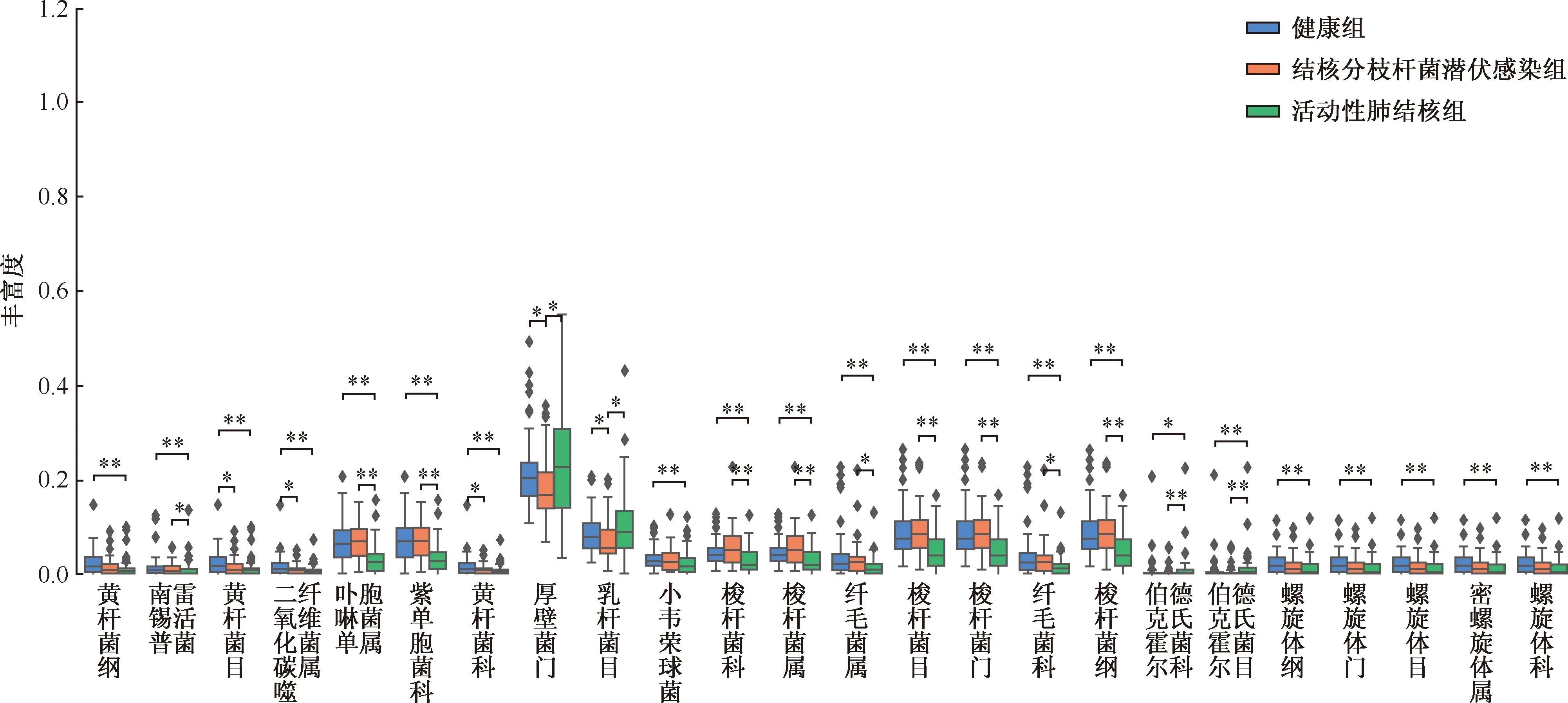
图中星号表示组间丰富度差异有统计学意义(*P<0.05,**P<0.01)
讨 论
既往研究认为,健康人的肺部是无任何细菌的,但随着高通量测序技术的发展,16S rRNA基因测序技术在细菌的鉴定与分类研究中发挥着越来越重要的作用,并发现健康人的肺部存在多种微生物[6]。16S rRNA基因普遍存在于细菌细胞,在细菌基因组中位于核糖体小亚基,含有10个保守区域(conserved regions)和9个高变区域(variable regions),具有良好的进化保守性和适宜的分析长度(约1540 bp),以及与进化距离相匹配的良好变异性。因此,16S rRNA被认为是细菌系统发育学研究和细菌分子鉴定的标准标识序列[13-14]。
本研究发现,健康人群、LTBI者和活动性肺结核患者的OTU数目无明显差别。通过Alpha多样性分析可知,MTB感染未改变人体痰液样本的物种丰富度,但是却造成活动性肺结核患者痰液样本中物种均匀度的变化。2012年,Cui等[15]发现结核病患者痰液微生物群比健康人更多样化,与笔者研究结果存在差异。菌群结构组成分析显示,门水平上,健康人群、LTBI者和活动性肺结核患者痰样本中的优势菌群均为拟杆菌门(Bacteroidetes)、变形菌门(Proteobacteria)、厚壁菌门(Firmicutes)和梭杆菌门(Fusobacteria),只是在各组中相对丰度有所变化。活动性肺结核患者痰样本中,相对丰度较高的前4位菌属分别为奈瑟菌属(Neisseria)、普氏菌属(Prevotella)、链球菌属(Streptococcus)和韦荣球菌属(Veillonella)。而2016年Krishna等[16]研究表明,链球菌属(Streptococcus)、奈瑟菌属(Neisseria)和韦荣球菌属(Veillonella)是印度结核病患者痰液菌群的3个优势属,他们还发现,厚壁菌门(Firmicutes)和放线菌门(Actinobacteria)是结核病患者的主要门,而拟杆菌门(Bacteroides)和变形杆菌门(Proteobacteria)在对照组(健康人群)中较高[16]。另外,2013年Cheung 等[17]比较了结核病患者和具有结核病类似咳嗽症状但痰菌培养阴性的对照人群之间的痰液菌群差异,发现这两类人群的菌群多样性类似,放线菌属(Actinomyces)、梭杆菌属(Fusobacterium)、纤毛菌属(Leptotrichia)、普氏菌属(Prevotella)、链球菌属(Streptococcus)和韦荣球菌属(Veillonella)是结核病患者痰液菌群的核心菌属。这三项研究表明,不同地域的结核病患者痰样本中优势菌群可能存在差异性。
不同分类水平上菌群组间差异物种分析发现,活动性肺结核患者痰样本中伯克霍尔德菌目(Burkholderiales)和伯克菌科(Burkholderiaceae)的相对丰度较健康人群和LTBI者明显增加。因为慢性排斥反应,肺移植者的长期存活率很低,2013年Borewicz等[18]研究发现,肺移植者的肺部菌群与健康人最明显的差异是伯克菌科(Burkholderiaceae)的出现。青枯菌属(Ralstonia)、贪铜菌属(Cupriavidus)和已知的肺部病原体伯克菌属(Burkholderia)都属于伯克菌科(Burkholderiaceae)。而青枯菌属(Ralstonia)[19-20]、苍白杆菌属(Ochrobactrum)[21]、细杆菌属(Microbacterium)[22-23]和贪铜菌属(Cupriavidus)[21]均与呼吸道感染、菌血症和院内感染有关。正常情况下,微生物菌群处于动态平衡状态,调节宿主的免疫反应,参与能量代谢,维护机体内环境稳定。它们在移植患者肺部的存在,与它们在疾病中发挥潜在作用或使有效的肺清除机制缺乏是一致的。现已发现,失衡的肺部菌群对免疫细胞亚群数量及功能的干扰、紊乱失调的免疫细胞弱表达或不表达重要病原体识别受体和结核毒性细胞因子、调节正常免疫功能的菌种被削弱或耗尽、正常菌群代谢产生的短链脂肪酸和吲哚丙酸减少等可能是导致结核病的致病机制[24]。
综上所述,本研究通过比较健康人群、LTBI者和活动性肺结核患者痰液菌群结构的变化,发现了三者在不同分类水平上具有明显差异的物种。但是由于本次研究对象数量较少,研究结果具有一定的局限性,后续还需扩大研究对象数量以进一步探索和明确健康人群、LTBI者和活动性肺结核患者的菌群特征,了解MTB感染对呼吸道菌群的确切影响。
猜你喜欢
今日农业(2022年14期)2022-09-15
中老年保健(2022年2期)2022-08-24
中国饲料(2022年5期)2022-04-26
昆明医科大学学报(2022年3期)2022-04-19
中国CT和MRI杂志(2021年12期)2021-11-25
海峡姐妹(2018年4期)2018-05-19
新课程·中学(2017年7期)2017-08-15
初中生学习·高(2016年8期)2016-05-14
中国民族民间医药·下半月(2014年2期)2014-09-26
浙江中医杂志(2004年5期)2004-03-09
