
LINC00092介导谷氨酸的兴奋性毒性在肝性脑病中的作用机制
2021-01-22 03:26李锦忠杨阮阮李敏然龚晓兵
暨南大学学报(自然科学与医学版) 2021年1期
李锦忠, 杨阮阮, 李敏然, 龚晓兵
(暨南大学 附属第一医院 感染科, 广东广州 510632)
肝性脑病(hepatic encephalopathy,HE)是肝脏功能障碍引起的中枢神经系统(central nervous system,CNS)功能失调综合征,其中一个主要的病因是肝代谢功能下降导致的毒物聚集,当肠源性毒性产物透过血脑屏障后,在脑组织中积聚,从而影响神经递质系统,最终导致患者出现神经功能障碍,如早期的兴奋性增高,以及晚期的意识障碍、行为失常和昏迷等[1].如何早期识别HE的神经系统功能障碍,预防不可逆的脑损伤是HE治疗的关键环节之一.HE发病机制复杂,主要学说有氨中毒、代谢紊乱、氨基酸失衡及氧化应激等[2].星形胶质细胞是血脑屏障的关键成分,通过维持脑中的溶质水稳态来保护神经元免受兴奋性毒性.因此,星形胶质细胞紊乱成为脑水肿和神经元改变的关键特征[3].
谷氨酸(glutamate,Glu)是CNS中含量最丰富的兴奋性神经递质,而HE的发生与氨过多、神经递质功能失衡有关[4].HE患者及其动物模型中Glu神经递质功能不良,其受体结合位点减少甚至缺失[5].所以HE患者的血氨影响Glu受体功能与代谢,进而导致HE患者的脑细胞功能紊乱,但具体机制不明.
长链非编码RNA(long noncoding RNA, LncRNA)是长度大于200个核苷酸的非编码RNA.研究表明LncRNA在剂量补偿效应、表观遗传调控、细胞周期调控和细胞分化调控等众多生命活动中发挥重要作用,成为遗传学研究热点[6-7].LncRNA在缺血缺氧性脑病、脑胶质瘤、精神疾病和脑发育等方面的研究不断有新的发展和突破,但有关LncRNA对肝性脑病的作用和调控机制知之甚少.本研究小组首次发现一个在HE中特异性高表达的LncRNA—LINC00092,对其进行初步鉴定:LINC00092全长3064bp,利用编码潜力评估工具 (coding potential assessment tool,CPAT)对其编码能力进行预测,结果提示无编码能力.Zhao等[8]发现卵巢癌细胞旁分泌诱导LINC00092过度表达,LINC00092与PFKFB2相互作用,在卵巢癌中诱导糖酵解表型并促进卵巢癌转移.Huang等[9]也证实LINC00092在结直肠癌中的诊断和预后能力.因此,探索LINC00092在HE的发生发展中的作用,对于寻找合理有效的HE预防和治疗措施具有十分重要的意义.
1 材料和方法
1.1 筛查HE中起关键作用的LncRNA
本研究小组从基因表达数据库(gene expressionomnibus,GEO) (https://www.ncbi.nlm.nih.gov/geo/)下载了芯片GSE57193的数据,包括4个肝硬化伴HE和4个健康对照(healthy control,HC)的脑组织样本的基因表达谱,根据P<0.05和|log2FC|>0.5的标准,利用R(3.6.1)语言(https://www.r-project.org/)的limma包筛选出HE和HC中差异的LncRNA和mRNA(图1).
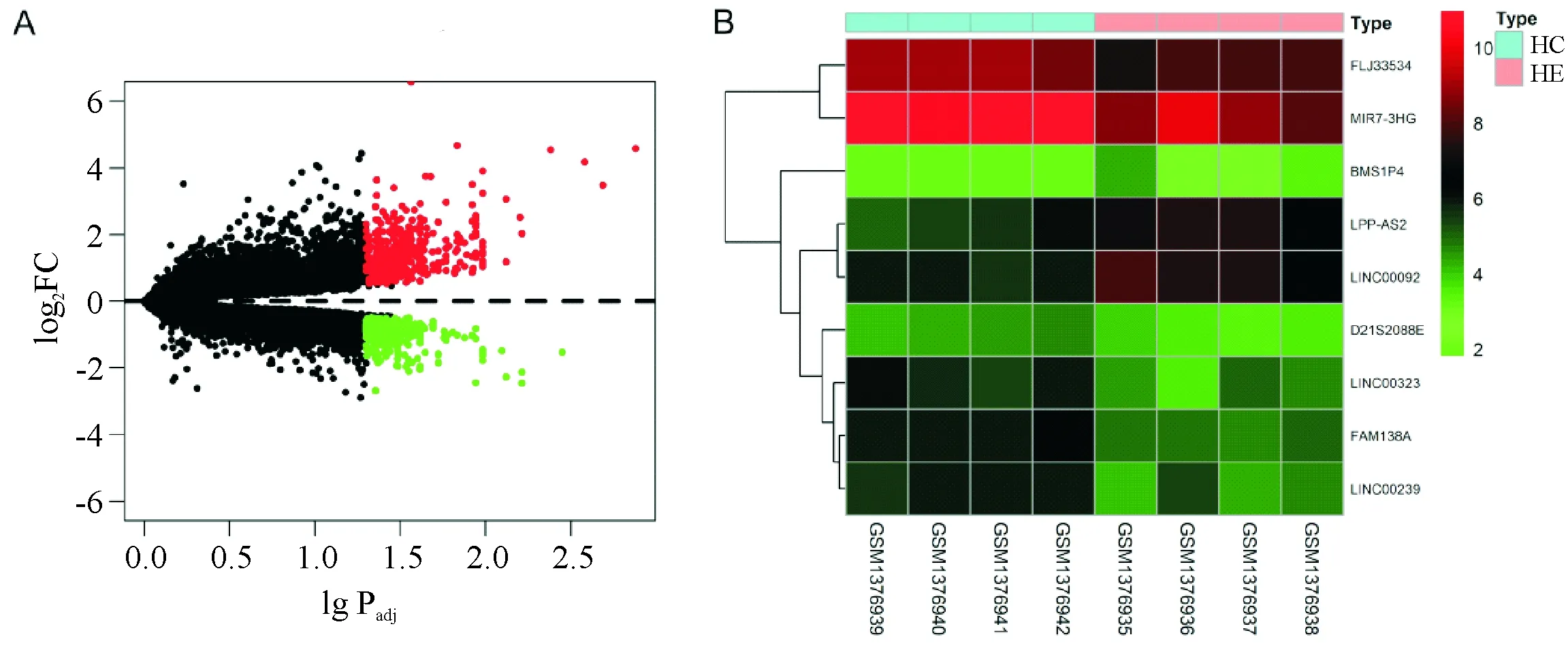
A.log2FC代表取对数后差异表达基因调整倍数(0.5倍),-lgPadj代表校正后的P值(0.05),图中的红点表示上调的差异表达基因,绿点代表下调的差异表达基因,黑点代表差异改变不明显的基因.B.顶端粉红色代表HE,蓝色代表HC;红色表示该基因在样品中高表达,而绿色在样品中低表达.
1.2 KEGG信号通路分析
根据差异表达基因的符号和序列号(ID),利用clusterProfiler包进行差异表达基因的京都基因和基因组百科全书(kyoto encyclopedia of genes and genomes,KEGG)分析,以P<0.05为标准输出结果.
1.3 ceRNA网络的构建
竞争性内源RNA(competitive endogenous RNA,ceRNA)网络的构建方法如下:从miRcode数据库下载差异表达LncRNA靶向的miRNA数据,使用Targetscan、miRdb和miRTarBase预测miRNA靶向的mRNA,然后与差异表达mRNA取交集得到目标mRNA.最终保留与差异表达的lncRNA,miRNA和mRNA的交叉点.Cytoscape(3.72)用于构建lncRNA-miRNA-mRNA ceRNA网络.
1.4 LINC00092在肝性脑病中机制假说的预测
通过RNA结合蛋白特异性数据库 (the database of RNA-binding protein specificities,RBPDB) (http://rbpdb.ccbr.utoronto.ca/)预测LINC00092的RNA结合蛋白(RNA-binding proteins,RBP),根据表达的相关性和RNA结合蛋白,提出LINC00092在肝性脑病中的机制假说.
2 结果
2.1 差异表达的LncRNA和mRNA及KEGG富集分析
根据P<0.05和|log2FC|>0.5的标准,从正常脑组织中筛选出HE中9个特异的LncRNA和728个特异的mRNA,其中LINC00092相对HC在HE中特异性高表达(图1).差异表达基因的KEGG富集分析显示,它们主要集中在脂肪酸降解、过氧化物酶体增殖剂激活受体(peroxisome proliferators-activated receptors,PPAR)信号通路、矿物质的吸收、缬氨酸、亮氨酸和异亮氨酸的降解、β-丙氨酸新陈代谢、脂肪酸代谢通路上(表1).

表1 差异表达基因的KEGG通路富集分析Table 1 Enrichment analysis of differentially expressed genes by KEGG pathway
2.2 LINC00092在肝性脑病中的作用机制
通过ceRNA网络(图2)可以看到LncRNA可通过竞争结合共同的micRNA应答元件(microRNA response elements,MREs)实现调节mRNA的表达.根据ceRNA网络和RBP(表2),发现LINC0009在HE中存在3个调控通路:(1)LINC00092/hsa-miR206/GJA1轴介导Glu的神经兴奋毒性损伤;(2)LINC00092/hsa-miR184/LRRC8A轴介导星形胶质细胞的溶胀激活和ATP诱导的兴奋性氨基酸释放;(3)LINC00092-A2BP1轴介导的Glu再摄取(图3).
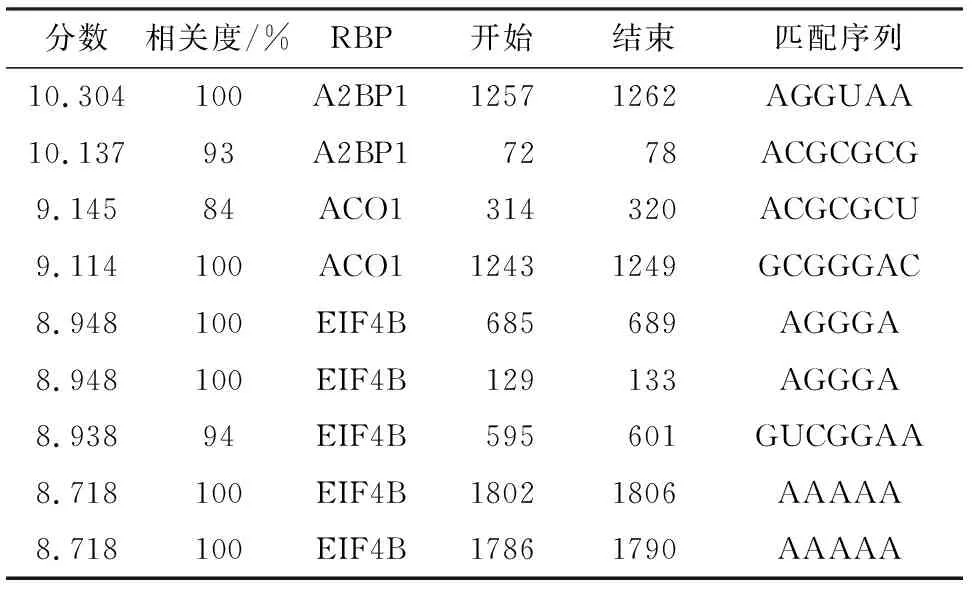
表2 LINC00092与多个RNA结合蛋白存在结合靶点
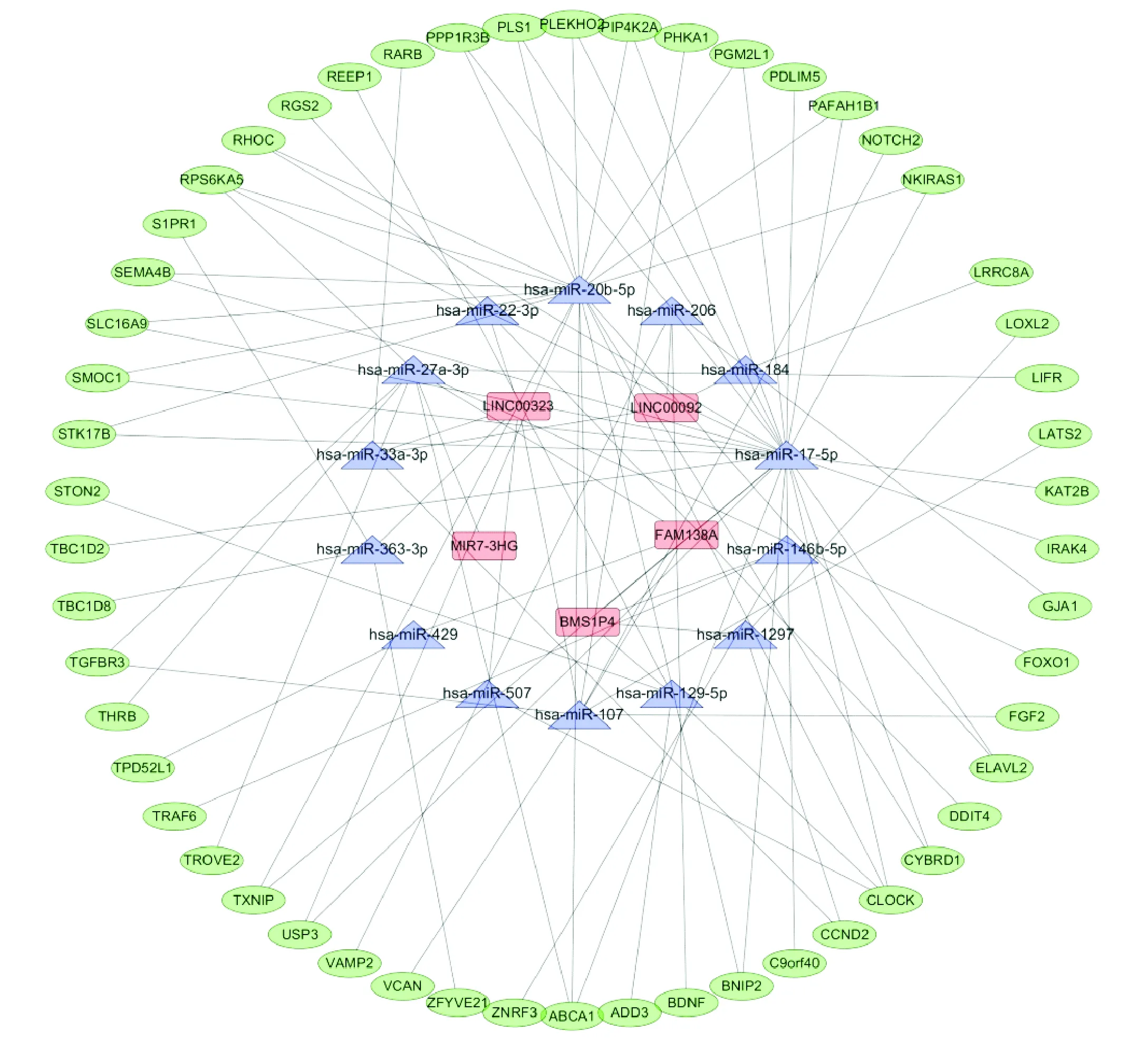
红色代表的是LncRNA,蓝色代表的是miRNA,绿色代表的是mRNA,任何2个基因间的连线代表两者间的相互作用.
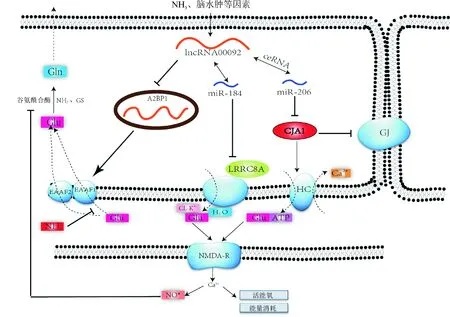
图3 LINC00092介导谷氨酸的兴奋性毒性在肝性脑病中的机制假说
3 讨论
目前LncRNA在肝性脑病的研究仍是空白,本研究小组首次发现一个在HE中特异性高表达的LncRNA—LINC00092,它的编码基因位于9号染色体reverse链96019724至96027993号碱基处,共6个转录本,本研究的研究对象为LINC00092-202,全长3064bp,结果发现LINC0009在HE中存在3个调控路径,各个路径间存在相互作用.
缝隙连接(gapjunction,GJ)是间隙连接通道的簇,能介导电信号和化学信号有效且快速的双向细胞间传输,半通道(hemichannel,HC)是由GJA1[也称为连接蛋白(connexin,Cx)43],组成的六聚体结构,介导细胞内外的信息交流[10-11]. LINC00092/hsa-miR206/GJA1轴介导Glu的神经兴奋性毒性损伤.GJA1是连接蛋白家族中在脊椎动物中高度保守的成员,以六聚体复合物的形式存在于质膜上,并起连接蛋白半通道的作用,从而发挥调控小分子和离子流通的功能[12]. GJA1是星形胶质细胞中含量最多的连接蛋白,而星形胶质细胞在正常的神经活动中起主要作用,它们为神经元提供代谢和结构支持,控制细胞外谷氨酸、钾、氢离子浓度,调节细胞外液的容量[13-15].这些功能取决于相邻两个星形胶质细胞通过GJ形成的功能合胞体,对谷氨酸、钾等起到“空间稀释”作用[16].当受到外来侵犯时,受损细胞通过GJ将Glu等有害物质释放到相邻细胞,起到了自我保护作用.同时GJA1还可作为HC参与旁分泌交流[17],但这种活动通常与病理状况有关[18].HC通道的开放导致大量Glu、ATP等毒性物质的释放,Glu通过兴奋性毒性杀死神经元,一方面是Glu下游的N-甲基-D-天冬氨酸(N-methyl-D-aspartic acid,NMDA)受体的持续活化以及随后大量Ca2+涌入有活力的神经元引起的兴奋性毒性损伤的主要信号转导剂,它可能通过各种机制进入细胞,但最重要的机制是通过与NMDA受体偶联的离子通道进入细胞[19].其次是由于HC通道是通过Ca2+内流介导的Glu释放,二者都使细胞内线粒体Ca2+超载,细胞膜去极化而导致神经元功能障碍和细胞死亡[20],同时活性氧的产生可以抑制星形胶质细胞谷氨酰合酶的活性,干扰谷氨酸-谷氨酰胺循环[21].本研究发现LINC00092、hsa-miR-206和GJA1之间存在调控关系,其中LINC00092和GJA1呈共表达关系,提示LINC00092可能通过吸附hsa-miR-206靶向调控下游GJA1,抑或通过调控hsa-miR-206的前体转录本的水平.但病理状态下表达上调的GJA1是通过何种机制关闭GJ通路和开放HC通路仍不清楚.介导GJA1在这两种通路间的转变可成为一种新的治疗方向.
LINC00092/hsa-miR184/LRRC8A轴介导星形胶质细胞的溶胀激活和ATP诱导的兴奋性氨基酸释放.LRRC8A(leucine-rich repeat-containing protein 8A)属于Lrrc8基因家族,其他成员包括Lrrc8b,Lrrc8c,Lrrc8d,和Lrrc8e[22-23].体积调节阴离子通道(the volume-regulated anion channel,VRAC)通过细胞膨胀激活,并在细胞体积调节中起关键作用.VRAC在脊椎动物细胞中普遍表达,并且还与许多其他生理和细胞过程有关,包括液体分泌、Glu释放、膜电位调节、细胞增殖、迁移和凋亡[24].LRRC8A及4个其他同源物形成了异源VRAC通道,但连接LRRC8A跨膜结构域2和3的细胞内环(IL)和连接LRRC8C,LRRC8D或LRRC8E跨膜结构域1和2的第1个细胞外环(EL1)都是VRAC激活所必需的[25].细胞膨胀后,VRAC活化导致释放Cl-和有机渗透物(例如牛磺酸,谷氨酸和天冬氨酸)[26-28],这与细胞内K+的挤出平行[29].与此同时,离子流出会产生驱动水流出所需的渗透梯度,从而使生理细胞体积得以恢复[30].肝脏功能障碍引起毒物聚集,当肠源性毒性产物透过血脑屏障后,在脑组织中积聚引起脑水肿.结合前期研究推测:脑水肿刺激脑组织上调LINC00092的表达,LINC00092可能通过吸附hsa-miR-184靶向调控下游LRRC8A,抑或通过调控hsa-miR-184的前体转录本的水平.LRRC8A通过VRAC反馈性调控细胞体积的同时,也过量释放了Glu,后者可通过兴奋性毒性杀死神经元.
LINC00092-A2BP1轴介导的Glu再摄取.A2BP1(ataxin 2-binding protein 1),也称为 RBFOX1,是一种神经元特异性剪接因子,对替代剪接具有正调节作用和负调节作用[31-32].该基因突变已被证实与癫痫、智力发育迟滞[33-34]以及自闭症[35]等一系列神经发育障碍性疾病相关.Rbfox1通过与可选择性外显子两翼的内含子中的(U)GCAUG 序列结合,以达到调控神经系统选择性剪接作用的目的[36].RBFOX1的靶基因中包括了与癫痫和共济失调相关的谷氨酸转运蛋白 EAAT1[37].Gehman等[38]发现Rbfox1的基因缺失导致对自发和海藻酸诱发的癫痫发作的敏感性增加,电生理记录显示,基因敲除小鼠的齿状回中神经元兴奋性相应增加.同时全转录组分析确定了Rbfox1(-/-)脑中的多个剪接变化,其中包括GABA-A受体γ2、NMDA受体1.这些剪接变化改变了介导突触传递和膜兴奋的蛋白质.因此,A2BP1指导了预防神经元过度兴奋和癫痫发作所需的遗传程序,但具体机制不明.因此提出假设:HE状态下过表达的LINC00092结合A2BP1后,可将A2BP1定位至特定的DNA上,通过可变剪切下调EAAT1的转录,影响了EAAT1对突触间隙Glu的再摄取;同时与LINC00092结合的A2BP1对NMDA受体1可变剪切的调控能力减弱,使突触后膜上的NMDA受体1表达增多.以上两种情况加剧了LINC00092介导的Glu的细胞外蓄积及兴奋性毒性伤害.
综上,LINC00092在HE中特异性高表达,一方面通过半通道和VRAC介导Glu的释放,同时使细胞内线粒体Ca2+超载而造成神经元功能障碍和细胞死亡;另一方面LINC00092-A2BP1轴介导的谷氨酸-谷氨酰胺循环障碍,导致Glu的细胞外蓄积,两方面共同导致神经元的兴奋性毒性.
猜你喜欢
中国典型病例大全(2022年9期)2022-04-19
河南农业·综合版(2022年1期)2022-03-01
中国药学药品知识仓库(2021年18期)2021-02-28
保健文汇(2020年3期)2020-08-22
阅读(科学探秘)(2019年5期)2019-07-19
教育教学论坛(2019年18期)2019-06-17
分析化学(2019年3期)2019-03-30
中西医结合心血管病电子杂志(2018年28期)2018-11-19
中国医学创新(2016年33期)2017-02-28
家庭医学(2015年10期)2015-11-10
