
柱芳烃与烷烃间C―H…π和C―H…O的作用本质及协同性
2020-09-21 02:12:22王一波
高等学校化学学报 2020年9期
孙 涛,王一波
(贵州大学化学化工学院,贵州省高性能计算化学重点实验室,贵阳550025)
大环主体化合物的结构和主客体化学性质是超分子化学的研究重点. 2008 年Ogoshi 等[1]在研究Lewis酸催化下对苯二甲醚和多聚甲醛聚合反应时得到一种白色晶体副产物,单晶结构分析发现是由对苯二甲醚通过亚甲基桥在苯环的对位连接而成的环状低聚物,因其分子呈柱状结构,将其命名为柱芳烃. 柱[5]芳烃的内径约为0.47 nm,与葫芦脲[6]和α-环糊精的内径近似[2,3],且更易溶于有机溶剂的主体分子,在分子识别和超分子组装等方面的应用引起了广泛关注.Yang等[4,5]提出的基于柱芳烃的超分子组装诱导荧光发射增强,为超分子功能体系在传感检测、药物递送、生物成像、自组装和智能发光材料等领域带来了新的可能性;基于柱芳烃的分子尺度多孔材料可对特定小分子进行选择性识别和捕获[6],Yang等[7]首次将全羟基柱[5]芳烃和柱[6]芳烃超分子有机骨架材料用于CO2、N2、烃类气体的吸附和分离研究[8,9]. Huang等[10]合成带有直链烷烃的柱[5]芳烃,利用柱[5]芳烃与直链烷烃的相互作用自组装得到首例柱[5]芳烃的超分子聚合物[11],在此基础上设计制备了超分子聚合物、分子弹簧[12]等一系列超分子复合物. Li等[13~15]发现和发展了全烷基取代柱[5]芳烃与一系列取代烷烃中性客体分子的复合物,并认为C―H…O,C―H…π等分子间弱相互作用是其形成稳定络合物的主要作用. Ogoshi等[16]以全羟基柱[6]芳烃为原料制备了超分子有机框架材料,实现了对CO2、N2、正丁烷及一些有机蒸气的吸附;他们使用1H NMR 研究甲氧基柱[5]芳烃(MeP5)和桥联MeP5 二聚体与正烷烃CnH2n+2(n=6~12,16,20)的复合物,发现其能够与正烷烃形成主客体复合物,因MeP5的高度(0.90 nm)与正己烷的长度(1.00 nm)接近,认为正己烷是最合适的客体分子[17];Ogoshi 等[18]报道了乙基柱[5]芳烃(EtP5)在晶态下对烷基链长度和形状选择性吸附的开关行为,对正己烷的选择性吸附诱导了EtP5晶体的结构发生了转变,对正戊烷和正庚烷吸附中也发现了同样的现象,同时还发现低于4个碳的直链烷烃、环状或者支链的烷烃不可被EtP5吸附;在此基础上,Ogoshi等[18]又发现了EtP5在烷基链长度选择性吸附方面的应用,并在固液相中来分离不同链长的正烷烃. EtP5倾向于与较长的直链烷烃形成稳定的主客体复合物,Ogoshi等[19]认为由于烷基链和EtP5的苯环之间通过众多的C―H…π相互作用来决定选择性,使用分子力学方法计算3种不同作用模式的结合能,EtP5…CnH2n+2(n=6~16)以1∶1作用模式时,结合能在−85.35~−126.36 kJ/mol之间,C13H28后结合能增加很小.
现已有针对柱芳烃的理论研究,如Suvitha等[20]使用B3LYP-D3/def2-TZVP 方法计算MeP5与有机磷主客体复合物,结合能比较大,能够形成主客体复合物,分子静电势显示主客体复合物之间发生了电荷转移;他们还使用B3LYP-D3 方法计算了MeP5 与对乙酰基氨基酚复合物的结合能为−117.57 kJ/mol,吉布斯自由能为−22.09 kJ/mol,在室温下能够形成稳定的复合物[21]. Bhadane等[22]使用ωB97X方法计算1-氰基金刚烷与EtP5复合物,认为二者通过氢键和C―H…π形成主客体复合物.
正烷烃是重要的化学化工原料,因其没有官能团,且具有低极化和疏水性,很难与大环如环糊精、冠醚、杯芳烃和瓜环等形成主客体复合物. MeP5的静电势显示柱芳烃腔内苯环和端基氧区域电子较为聚集,正烷烃在MeP5的空腔内可以形成C―H…π键,而在两端也可形成C―H…O键,但尚不确定二者之间是否存在协同作用,因此,本文应用量子化学计算方法研究了MeP5 与CnH2n+2(n=1~10,12,14,16)分子间的相互作用,探讨了C―H…π与C―H…O相互作用本质以及协同性,并计算分析了超分子复合物的热力学函数.
1 计算方法
密度泛函理论在分子间弱相互作用中的应用较为广泛,Grimme的色散校正泛函DFT-D3[23]和DFTD3(BJ)[24]能够较准确地计算分子间非键相互作用;Head-Gordon等[25]发展的杂化泛函ωB97X-V在计算分子间非键相互作用也有较好的结果. MeP5…CnH2n+2(n=1~10,12,14,16)复合物的原子数为110~155,即使使用def2-SVPD[26]基组,基函数个数也在1582~2122之间,因此准确计算MeP5…CnH2n+2(n=1~10,12,14,16)复合物的分子间相互作用对密度泛函理论方法仍具有挑战性. 为了找到准确计算MeP5与CnH2n+2(n=1~10,12,14,16)分子间相互作用的密度泛函理论方法,将MeP5…C8H18复合物的柱芳烃进行裁剪,如图1所示,使用高精度的CCSD(T)/CBS[27][(kJ/mol)计算方法见式(1)]作为标准值,并与多种密度泛函计算理论方法作比较,探讨计算结果的可靠性(见表1).由MP2/CBS的结合能和高级电子相关校正项组成.
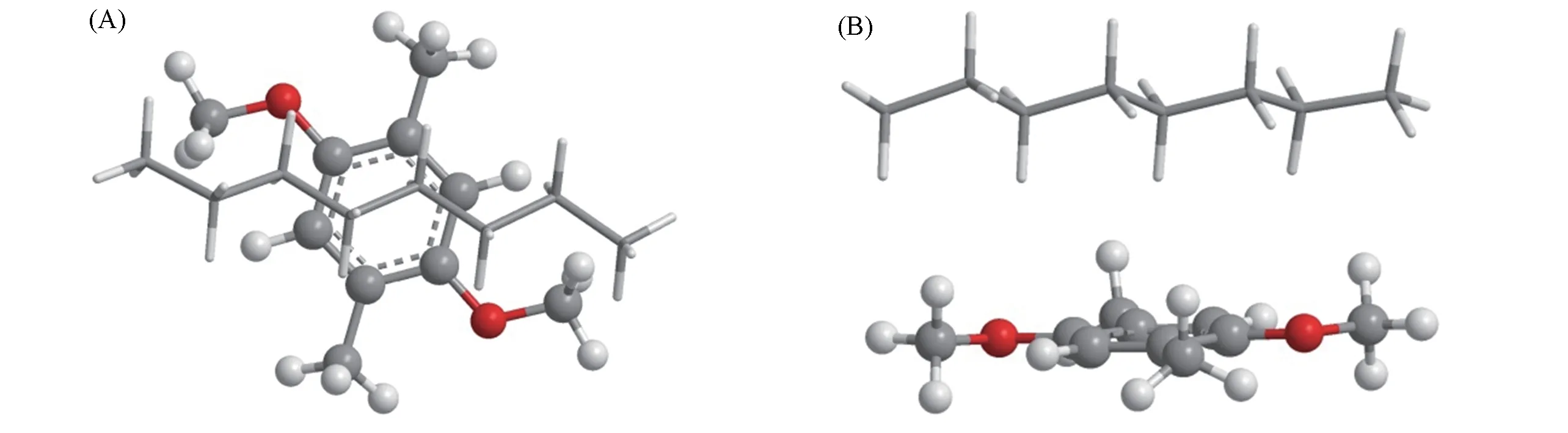
Fig.1 Top(A)and side(B)view of C10H14O2…C8H18
由表1可见,不论使用哪种密度泛函方法,def2-SVPD与def2-TZVPP基函数经BSSE校正后的计算结果相差不大. 使用ωB97X-D[28]方法计算的结合能比CCSD(T)/CBS 标准值约大4.26 kJ/mol,而B2PLYP-D3BJ[24]方法计算结果比CCSD(T)/CBS小2.22 kJ/mol;CAM-B3LYP-D3BJ[24]和M06-2X-D3[29]计算结果较为准确,CAM-B3LYP-D3BJ方法计算的结合能更接近CCSD(T)/CBS标准值;ωB97X-V等其它8种方法的2种基函数的计算结果增大了约0.5~1.5 kJ/mol. PW91系列泛函是计算热力学函数使用较多的方法之一,我们[30]研究发现,B3PW91-D3BJ方法在计算热力学函数方面具有高可靠性,因此选择B3PW91-D3BJ[24]/def2-SVPD 计算几何结构和热力学函数,使用CAM-B3LYP-D3BJ/def2-SVPD 计算其结合能. 结合能计算均采用Boys-Bernardi完全均衡校正法(CP)进行基函数重叠误差(BSSE)校正[31].
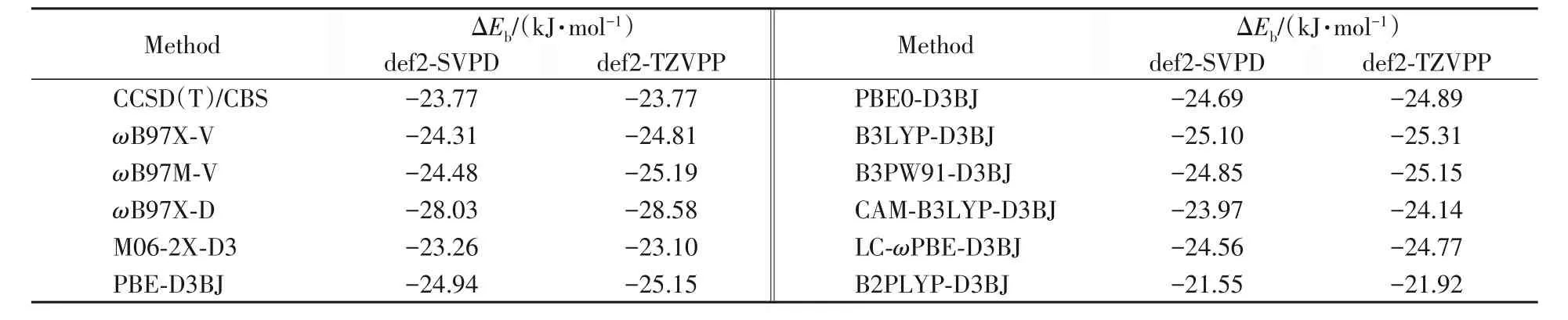
Table 1 Binding energies(ΔEb)of C10H14O2…C8H18complex using density functional theory methods with different basis sets
使用二代绝对局域分子轨道能量分解分析(ALMO-EDA)方法[32,33]对柱芳烃与正烷烃分子间相互作用进行能量分解,该方法将分子间结合能分解为冻结密度(Frz)项、极化项(Pol)和电荷转移(CT)项.冻结能量项进一步分解为静电能ΔEElec(kJ/mol),Pauli排斥能ΔEPauli(kJ/mol)和色散能ΔEDisp(kJ/mol).

所有计算均采用Gaussian 16[34]程序和Q-Chem[35]程序完成.
2 结果与讨论
2.1 几何结构和结合能分析

Fig.2 Top(A)and side(B)views of optimized geometries of MeP5

Fig.3 Top(A)and side(B)views of optimized geometries of MeP5…C4H10
使用B3PW91-D3BJ/def2-SVPD 方法对MeP5(图2)和MeP5…CnH2n+2(n=1~10,12,14,16)复合物(图3)的几何结构进行全自由度能量梯度优化,对平衡几何结构进行振动频率计算,复合物无虚频,表明几何结构为对应势能面的极小点. 计算结果表明,正烷烃分子在MeP5的空腔内主要形成C―H…π键,而在两端形成C―H…O键. 在MeP5…CH4复合物中,CH4在MeP5空腔中,C―H到MeP5平面的距离分别为0.35,0.27 和0.27 nm(图S1,见本文支持信息),由于CH4分子较小,二者强相互作用使MeP5的几何结构已经发生形变;C2H6与MeP5强相互作用也使得C2H6与MeP5形成复合物时几何结构发生了形变. 随着正烷烃分子长度的增加,复合物的稳定性也在增加. 在MeP5…C3H8复合物中,C3H8分子在柱芳烃的中间位置,C3H8中间C―H到MeP5柱平面的距离为0.27和0.26 nm(图S2,见本文支持信息),在MeP5…C3H8复合物中存在两种类型的C―H…π键,一种是C―H与MeP5苯环形成C―H…π键,另一种是C3H8两端C―H指向柱芳烃苯环的C=C键位置. C3H8的端基C原子到O原子最小距离为0.40 nm,C―H 到O 原子最小距离为0.31nm,能形成C―H…O 键;在MeP5…C4H10复合物中,C4H10的一端C 原子到O 原子距离分别为0.40 和0.42 nm,C―H 到O 原子的距离均为0.31 nm,也能形成C―H…O 键,如图3 所示. 随着C 原子数的增加,烷烃分子的长度也逐渐增加,MeP5 空腔的长度为0.87 nm,C6H14(分子长度0.82 nm)以后的烷烃分子逐渐延伸到MeP5空腔外(图S3,见本文支持信息).
由表2 可见,B3PW91-D3BJ/def2-SVPD 方法计算的结合能比CAM-B3LYP-D3BJ 大约13%(MeP5…CH4,7.75%). MeP5…CnH2n+2(n=1~10,12,14,16)复合物的结合能较大,在−30.33~−126.98 kJ/mol 之间,随着正烷烃分子C 原子数的增加,结合能也在逐渐增大. 当正烷烃CnH2n+2(n=1~4)分子长度较短时,复合物的结合能随着碳链的增长,结合能增加较大,高达−15 kJ/mol 以上.正烷烃CnH2n+2(n=1,2)分子长度较短,与柱芳烃形成复合物后,柱芳烃的几何结构已发生形变;在MeP5…CnH2n+2(n=3,4)复合物中,正烷烃已与柱芳烃端口的甲氧基存在相互作用,能够形成C―H…O键. 正烷烃CnH2n+2(n=5~8)与柱芳烃形成的复合物在空腔内不仅存在C―H…π键,在柱芳烃的端口还能形成多个C―H…O键,此时随着碳链的增长,结合能增加约−6~−10 kJ/mol. 正烷烃CnH2n+2(n=9,10,12,14,16)分子长度较长(>1.20 nm),已延伸到MeP5 空腔外,随着C 原子数的增加,结合能的增加<−4 kJ/mol. 在复合物中,结合能随着烷烃分子碳链的增长而逐渐增加,而增加幅度逐渐减小.
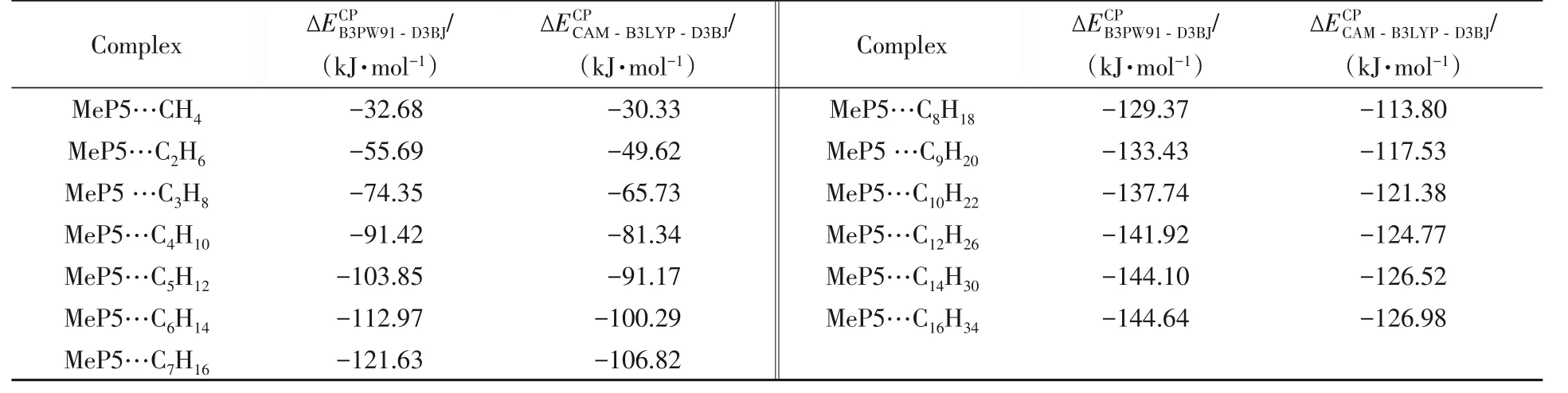
Table 2 Binding energies of MeP5…CnH2n+2(n=1―10,12,14,16)
Ogoshi等[19]设计了1∶1作用模型、鱼骨状模型和2∶1的一维通道结构3种简单作用模型,通过分子力学方法计算结合能. 1∶1 作用模式中,EtP5…CnH2n+2(n=6~16)的结合能在−85.35~−126.36 kJ/mol 之间,C13H28后结合能增加很小,几乎不再变化. 而2∶1作用的一维通道结构结合能呈线性增加. 由表2可见,使用分子力学计算二者结合能的结果可能偏小;在C10H22以后,正烷烃碳链的增长结合能增加很小,与Ogoshi等[19]的结论一致,C10H22与C13H28有差别可能是因为所选用的MeP5高度小于EtP5.
由C6H14形成复合物前后的静电势图(图4)可见,在C―H…π和C―H…O 作用区域静电势数值明显变小,目前还无法将MeP5…CnH2n+2(n=1~10,12,14,16)复合物中C―H…π与C―H…O 完全分离,为探讨两种相互作用对其结合能和稳定性的影响,以MeP5…C12H26为例设计两种作用模型:模型A中,固定MeP5 结构不变,对C12H26端口片段(C4H10)进行部分优化,模型A 中分子之间主要是端口的C―H…O 作用;模型B 中,使O 向上翻转90°,使分子间主要为C―H…π作用,如图5 所示. 使用CAM-B3LYP-D3BJ/def2-SVPD方法计算了结合能,并进行能量分解,结果列于表3.
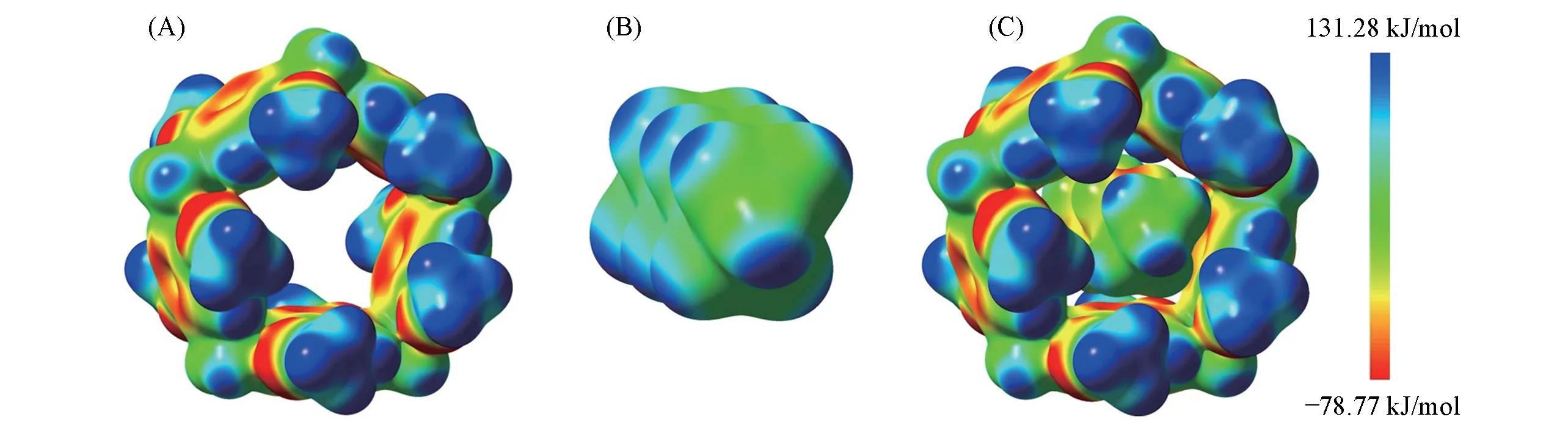
Fig.4 Electrostatic potential of MeP5(A),C6H14(B)and MeP5…C6H14(C)

Fig.5 Geometries of MeP5…C12H26(A),models A(B)and B(C)
由表3 可见,复合物中两端的C―H…O 作用能为−42.36 kJ/mol,C―H…π作用能为−72.25 kJ/mol,二者之和比MeP5…C12H26的结合能(−124.75 kJ/mol)小10.14 kJ/mol. 可见在MeP5…CnH2n+2(n=3~10,12,14,16)复合物中,C―H…π和C―H…O 是一种协同作用,对复合物的稳定性起重要作用.

Table 3 Binding energies and energy components of MeP5…C12H26 in models A and B*
为了详细研究MeP5与正烷烃分子间相互作用,在MeP5…C4H10复合物中,以C4H10中间两碳原子的中点为原点坐标,平行移动C4H10分子,使其逐渐远离MeP5空腔,使用CAM-B3LYP-D3BJ/def2-SVPD方法计算其结合能,结合能变化如图6 所示. 在C4H10向端口移动过程中,结合能逐渐减小,在MeP5端口(平移0.35 nm),二者之间有C―H…π和C―H…O作用,但是正烷烃与MeP5端口的―CH3的排斥作用增强[C4H10与MeP5 端口的―CH3最小距离仅为0.15 nm,图S4(见本文支持信息)],此时结合能最小,仅有−1.38 kJ/mol;继续平移C4H10,C―H…π逐渐消失,C4H10逐渐移至MeP5 空腔端口,同时正烷烃与MeP5端口的―CH3的排斥作用减小,C―H…O起主要作用,所以结合能又逐渐增大.

Fig.6 Binding energy of MeP5…C4H10 complexes at different distances
2.2 热力学函数计算
为了更进一步研究MeP5…CnH2n+2(n=1~10,12,14,16)的结合过程,使用B3PW91-D3BJ/def2-SVPD计算复合物的吉布斯自由能变(ΔG)和焓变(ΔH). Elm等[36]将ΔG分为两部分[ΔGbind=ΔEbind+ΔGthermal,ΔEbind(kJ/mol)和ΔGthermal(kJ/mol)分别表示结合能和热力学对Gibbs自由能的贡献]. ΔEbind的准确性直接影响ΔG,在计算过程中通常不考虑BSSE对ΔG的影响,但在DFT计算过程中BSSE对ΔEbind又不能忽略,因此使用DFT-C[37](B3PW91-D3BJ/def2-SVPD)方法进行BSSE计算. 计算结果见图7.
在MeP5…CnH2n+2(n=1,2)复合物中,298.15 K 下,101325 Pa 下,ΔG分别为1.52 和−2.65 kJ/mol,这可能与二者形成复合物使柱芳烃的几何结构发生了形变有关. 由图7可见,MeP5…C3H8的ΔG随着温度的升高变化较为平缓,并与MeP5…CnH2n+2(n=4,5)出现交叉,MeP5…C3H8的结合能相对较小,但是C3H8在MeP5的空腔内,需要较高的温度MeP5…C3H8才能释放C3H8. ΔG的绝对值随着温度的升高而逐渐减小,低温下二者形成复合物能够自发进行,放出热量,焓为主要驱动. ΔG,ΔH与结合能ΔE变化趋势基本一致,均呈现随着正烷烃链长的增加而逐渐增大的趋势.
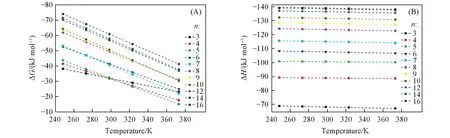
Fig.7 Gibbs free energy change(ΔG)(A)and enthalpy change(ΔH)(B)of MeP5…CnH2n+2(n=3―10,12,14,16)
2.3 ALMO-EDA计算
为了研究MeP5…CnH2n+2(n=1~10,12,14,16)空腔内C―H…π和端口C―H…O 协同作用的本质,使用ALMO-EDA 对正烷烃与MeP5 相互作用进行能量分解分析,结果见表4. ALMO-EDA 计算结果表明,C―H…π和C―H…O协同作用中静电能、色散能、极化和电荷转移都是吸引作用,而Pauli排斥能是排斥作用,对于MeP5…CnH2n+2(n=1~10,12,14,16)复合物,静电能和色散能起主导作用,约占总吸引作用的94%,色散能略大于静电能;极化项和电荷转移能仅占总吸引相互作用的6%左右. 在MeP5…CnH2n+2(n=1~4)复合物中,烷烃分子主要在柱芳烃空腔内,色散能达到50%左右,CnH2n+2(n=3,4)在MeP5 的端口已能形成C―H…O 键. 在MeP5…CnH2n+2(n=5~8)复合物中,随着烷烃链的增长,因烷烃与柱芳烃能够形成多个C―H…O 键,静电作用略有增强,但是色散作用依然略大于静电作用. 在MeP5…CnH2n+2(n=9,10,12,14,16)复合物中,烷烃两端在柱芳烃外面,碳链增长,静电作用和色散作用占总的吸引作用的比例基本不变. 由图8可见,在MeP5…CnH2n+2(n=3~10,12,14,16)复合物中,结合能增加幅度随着烷烃分子的增长而逐渐减小,而各能量分解项先增大,在MeP5…C5H12复合物达到最大后,又逐渐减小;同时Pauli排斥项增加幅度也是先增大后减小.

Table 4 ALMO-EDA energy components calculated with CAM-B3LYP-D3BJ/def2-SVPD*

Fig.8 Subtracted value between the binding ener⁃gy, electrostatics, dispersion, polarization and charge transfer of MeP5…CnH2n+2 and MeP5…Cn-1H2n(n=2―10,12,14,16)
综上所述,采用密度泛函理论B3PW91-D3BJ/def2-SVPD 方法优化了MeP5…CnH2n+2(n=1~10,12,14,16)复合物的几何结构,并用CAM-B3LYP-D3BJ/def2-SVPD 方法计算了复合物的结合能. 结果表明,随着烷烃分子碳链的增长,结合能逐渐增大;在298.15 K,101325 Pa下,MeP5与CnH2n+2(n=3~10,12,14,16)形成复合物的过程中,ΔG和ΔH均小于零,是自发进行的,且焓是主要驱动力. 在MeP5空腔内与烷烃主要存在C―H…π,在端口也可形成 C―H…O. 二者的协同作用对复合物的稳定性起到了重要作用;ALMO-EDA能量分解分析结果表明,C―H…π和C―H…O协同作用中静电能和色散能是其主要吸引作用,二者加和约占总吸引作用的94%,而极化能和电荷转移能仅占总吸引作用的6%.
支持信息见http://www.cjcu.jlu.edu.cn/CN/10.7503/cjcu20200291.
猜你喜欢
中学生数理化·八年级物理人教版(2023年10期)2023-11-30 01:58:12
中学生数理化·八年级物理人教版(2022年10期)2022-11-10 09:42:30
大学物理(2022年9期)2022-09-28 01:10:52
中学生数理化·八年级物理人教版(2021年10期)2021-11-22 08:00:10
西藏艺术研究(2021年3期)2021-06-02 09:36:46
马克思主义哲学研究(2020年2期)2020-07-21 01:36:26
物理通报(2020年7期)2020-07-01 09:28:02
中学生数理化·八年级物理人教版(2019年10期)2019-11-25 07:33:42
大观(2017年2期)2017-04-07 16:08:02
原子与分子物理学报(2015年3期)2015-11-24 12:49:34
