
Ce1−xMnxO2表面性质对催化CO2和甲醇直接合成DMC反应活性的影响
2020-09-21 02:12:30张国强孙宇辰史亚波郑华艳上官炬刘守军史鹏政
高等学校化学学报 2020年9期
张国强,孙宇辰,史亚波,郑华艳,李 忠,上官炬,刘守军,史鹏政
(1.太原理工大学煤科学与技术教育部和山西省重点实验室,2.化学化工学院,太原 030024;3.太原科瑞康洁净能源有限公司,太原 030006)
碳酸二甲酯(DMC)是一种环境友好且可生物降解的绿色化学品,可代替有毒的光气、氯甲烷和硫酸二甲酯等用于烷基化和羰基化反应[1]. DMC 的氧含量高达53%,且与汽柴油有良好的掺合性能,可以代替甲基叔丁基醚(MTBE)作为燃料添加剂. 另外,DMC还可作为锂离子电池的电解液[2]. 以CO2和甲醇为原料直接合成DMC(CO2+2CH3OH→CH3OCOOCH3+H2O)有利于促进CO2减排,并实现CO2资源化利用,是一条绿色的合成路线.
目前,催化CO2和甲醇反应合成DMC的催化剂种类众多[3~8]. 与均相催化剂相比,多相催化剂因便于分离、不易失活、易于循化和低毒环保等特点而被广泛研究. 在多相催化剂中金属氧化物具有制备方法简单和选择性高等优点,是催化CO2和甲醇合成DMC的理想催化剂. 其中,CeO2因其独特的表面性质和较高的催化活性成为近年来的研究热点.
首先,CeO2表面配位不饱和的Ce和O离子使其同时具有Lewis酸碱性[9,10]. 研究结果表明,CeO2表面碱性位吸附和活化CO2,随后与甲醇解离产生的CH3O*反应生成甲基碳酸酯(CH3OCO2*),另一分子甲醇在酸性位解离产生CH3*,并与CH3OCO2*反应生成DMC[11,12]. 另外,CeO2立方萤石结构中的Ce3+和Ce4+具有快速可逆转化的特征,使其表面容易产生一定数量的氧空位. 研究发现,CeO2表面氧空位能够增强对CO2分子的吸附和活化,有利于提高CO2和甲醇合成DMC的反应活性[13]. 向CeO2中掺杂适量的金属(如Zr,Ti,Ca,Al 或Zn)可显著提高催化剂表面酸碱性和氧空位含量,进而提高CO2和甲醇合成DMC的反应活性[14~18].
Mn 氧化物作为一种低成本催化剂在众多氧化还原反应中显示出良好的催化活性[19,20]. 另外,Mn作为助剂可掺杂进入CeO2晶格中形成固溶体,由于Ce与Mn之间的相互作用,使CeO2-MnOx固溶体显示出优越的氧化还原性能[21]. Mnn+掺杂进入CeO2晶格可代替一个Ce4+,造成电荷缺失和不平衡,促进氧空位生成,从而增强CeO2-MnOx固溶体对氧的吸收和释放性能[22]. Yao 等[23]研究发现,CeO2-MnOx固溶体表面酸性和氧空位数量明显高于纯CeO2,在低温NH3选择性还原NO 反应中显示出良好的催化活性. Dai等[24]研究发现,与纯CeO2相比,Mn掺杂形成的CeO2-MnOx固溶体表面碱性位数量显著增加,增强了催化剂对CO2的吸附,在水气变换反应中显示出优越的催化活性.
本文通过共沉淀法制备了不同Mn掺杂量的Ce1−xMnxO2催化剂,并在高压反应釜中对其催化CO2和甲醇合成DMC 的反应活性进行了评价,并研究了Ce1−xMnxO2表面酸碱性和氧空位含量对催化CO2和甲醇直接合成DMC反应活性的影响.
1 实验部分
1.1 试剂与仪器
Ce(NO3)3·6H2O(纯度≥99.0%),购于天津光复科技发展有限公司;NaOH(纯度≥96.0%)和Mn(NO3)2溶液(质量分数为49.0%~51.0%,分析纯),购于天津市风船化学试剂科技有限公司;甲醇(纯度≥99.9%)和无水乙醇(纯度≥99.5%),购于天津市科密欧化学试剂有限公司.
Rigaku D/max2500 型粉末X 射线衍射仪(XRD,日本Rigaku 公司);3H-2000PS1/2 型静态容量法比表面及孔径分析仪(北京贝世德公司);GG314-JEM-21000F型场发射透射电子显微镜(TEM,日本JEOL公司);Micromeritics AutoChem 2920型全自动程序升温化学吸附仪(CO2/NH3-TPD,美国Micromeritics公司);ESCALAB 250-Xi 型X 射线光电子能谱仪(XPS,英国Thermo Fisher Scientific 公司);ICP OES 730/Agilent 7700型等离子体发射光谱仪(ICP,美国安捷伦公司);GC-9160型气相色谱仪(上海欧华分析仪器厂).
1.2 实验过程
1.2.1 催化剂的制备 将3.475 g Ce(NO3)3·6H2O 放于30 mL 去离子水中并搅拌溶解. 取一定体积的Mn(NO3)2溶液(质量分数按50%计)与Ce(NO3)3溶液混合均匀,使混合液中Mn/(Ce+Mn)的理论摩尔比分别为0.025,0.05,0.10和0.15. 使用平流泵将上述溶液匀速滴加至2 mol/L的NaOH溶液中,持续搅拌. 随后将所得混合物在室温下陈化1 h后,过滤洗涤数次直至洗涤液呈中性. 将所得沉淀用乙醇离心洗涤3 次,然后置于80 ℃烘箱中干燥12 h. 将干燥后的固体充分研磨后,置于马弗炉中于550 ℃焙烧5 h得催化剂. 由等离子体发射光谱(ICP)表征测试得到催化剂中实际Ce和Mn元素含量,并将其命名为Ce1−xMnxO2,其中x代表Mn/(Ce+Mn)摩尔比. 称取2.864 g Mn(NO3)2溶液并稀释至30 mL,将其滴加至NaOH溶液中,其余步骤相同,所合成催化剂MnxOy作为空白对照.
1.2.2 催化活性评价 将0.2 g催化剂和30 mL 甲醇置于250 mL 内衬为聚四氟乙烯的高压反应釜中.首先,向反应釜中充入0.5 MPa的CO2气体,排空后再次充入0.5 MPa的CO2气体,重复3次将釜内空气吹扫出反应釜. 然后缓慢向反应釜中充入CO2气体至3 MPa,之后开始升温,当反应温度达到120 ℃时,反应压力上升至6.5 MPa,恒温反应4 h. 反应结束后取上层清液20 mL,向其中滴入300 μL正丙醇作为内标物,摇匀后静置,采用内标法对上层清液进行分析. 产物除DMC外未检测到其它物质,采用下式计算DMC的收率(mmol/g):
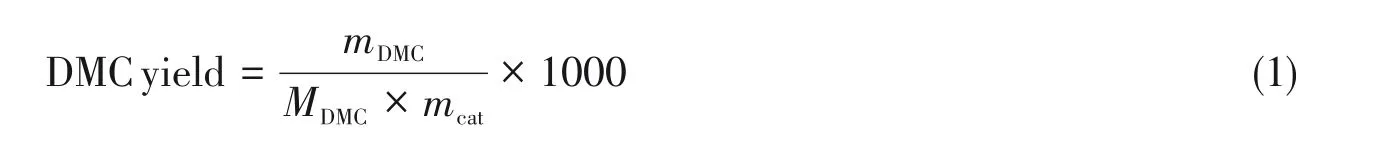
式中,mDMC(g)为产物中DMC的质量;MDMC(g/mol)为DMC的摩尔质量;mcat(g)为催化剂质量.
2 结果与讨论
2.1 催化剂的晶相结构分析
Ce1−xMnxO2催化剂的XRD 表征结果如图1所示. 可见,Ce1−xMnxO2催化剂均呈现明显的立方萤石结构(JCPDS No. 34-0394),表明Mn 掺杂并未改变CeO2的晶相结构. 其中2θ在28.5°,33.0°,47.5°,56.5°,59.4°和69.6°处出现的衍射峰可分别归属为CeO2的(111),(200),(220),(311),(222)和(400)晶 面[25]. 而 催 化 剂 中 并 未 检 测 到MnO2(JCPDS No.30-0820)和Mn2O3(JCPDS No.41-1442)的特征衍射峰. 通过分析发现,Mn掺杂后CeO2(111)晶面衍射峰向高角度方向偏移,表明形成了Ce-Mn-O固溶体[26]. 对Ce1−xMnxO2催化剂的晶胞参数和晶面间距进行分析,结果列于表1. 可见,Ce1−xMnxO2催化剂的(111)晶面对应的晶格常数和晶面间距均随Mn 掺杂量的增加而逐渐下降. 这是由于Mnn+离子半径(Mn2+离子半径为0.083 nm;Mn3+离子半径为0.0645 nm)明显小于Ce4+离子半径(0.097 nm),Mnn+进入CeO2晶格引起晶格收缩所致[27].

Fig.1 XRD patterns of Ce1-xMnxO2 catalysts

Table 1 Interplanar spacing,lattice parameters and crystallite size of Ce1−xMnxO2 catalysts
2.2 催化剂的织构性质分析
Ce1−xMnxO2催化剂的氮气吸附-脱附等温线如图2所示,催化剂在p/p0=0.6~1.0范围内均呈现明显的Ⅳ型吸附平衡等温线,且均具有H2型回滞环,表明催化剂均存在介孔结构.
催化剂的比表面积、孔径和孔体积等数据列于表2. 可以看到,催化剂Ce1−xMnxO2的比表面积均约为145 m2/g,Mn 掺杂后,除Ce0.956Mn0.044O2催化剂外,其余催化剂孔体积和孔径均略有增大. 但总体来说,Mn 掺杂并未显著影响Ce1−xMnxO2催化剂的织构性质.

Fig.2 N2 adsorption⁃desorption isotherms of Ce1-xMnxO2 catalysts

Table 2 Textural parameters of Ce1−xMnxO2 catalysts
2.3 催化剂的形貌分析
Ce1−xMnxO2催化剂的TEM照片如图3(A)~(E)所示. 可以看出,Ce1−xMnxO2催化剂均呈现无规则的纳米颗粒状,且纳米颗粒中夹杂着一定数量的短棒状纳米晶体.
CeO2形貌受温度、前驱体阴离子种类、沉淀剂类型和溶液pH的影响较大,该现象的出现可能是由于在催化剂制备的陈化过程中,Ce(OH)3晶核有序生长受温度、时间和pH的影响未能完全生长成为均一的纳米棒状晶体所致[28]. 通过对图中的大量纳米结构进行统计测量,分析了纳米颗粒的粒径分布和平均粒径[图3(A′)~(E′)]. 结果表明,催化剂颗粒尺寸分布在5~12 nm 之间,CeO2,Ce0.983Mn0.017O2,Ce0.956Mn0.044O2,Ce0.910Mn0.090O2,Ce0.864Mn0.136O2平均粒径分别为7.69,8.17,7.95,7.89 和7.85 nm. 可见,Mn掺杂并未对催化剂的颗粒尺寸和粒径分布造成显著影响[29].

Fig.3 TEM(A―E)images and particle size distributions(A′—E′)of Ce1-xMnxO2 catalysts
图4(A)~(E)示出了催化剂的HRTEM照片,可以看到,Ce1−xMnxO2催化剂均具有良好的晶体结构,且催化剂中均存在条纹间距为0.31 和0.27 nm 的晶格,分别对应于CeO2的(111)晶面和(200)晶面(JCPDS No.34-0394)[30,31]. Mn2O3(111)晶面的晶格条纹间距也为0.27 nm,然而XRD 表征结果显示催化剂中并未出现Mn 物种的特征衍射峰,因此条纹间距为0.27 nm 的晶格应对应CeO2的(200)晶面[32,33].
另外,并未发现CeO2的(110)晶面所对应的晶格条纹间距(0.19 nm),这可能是由于在高温下热处理的CeO2暴露晶面会向(111)晶面转化导致[31]. 主要原因是,在CeO2的所有晶面中,(111)晶面形成能最低,结构最稳定,(111)晶面暴露的数量在所有晶面中最多[34],这与XRD 表征结果一致. 催化剂的HRTEM照片中并未发现Mn氧化物的晶格条纹,表明Mnn+进入CeO2晶格中形成了固溶体[35,36].

Fig.4 HRTEM images of Ce1-xMnxO2 catalysts
2.4 催化剂的表面酸碱性分析
Ce1-xMnxO2催化剂的CO2-TPD和NH3-TPD曲线分别如图5(A)和(B)所示. 可见,催化剂表面均存在3种类型的酸性和碱性位,通过高斯拟合对催化剂表面酸碱性进行定量分析,结果列于表3.

Fig.5 CO2⁃TPD(A)and NH3⁃TPD(B)profiles of Ce1-xMnxO2 catalysts
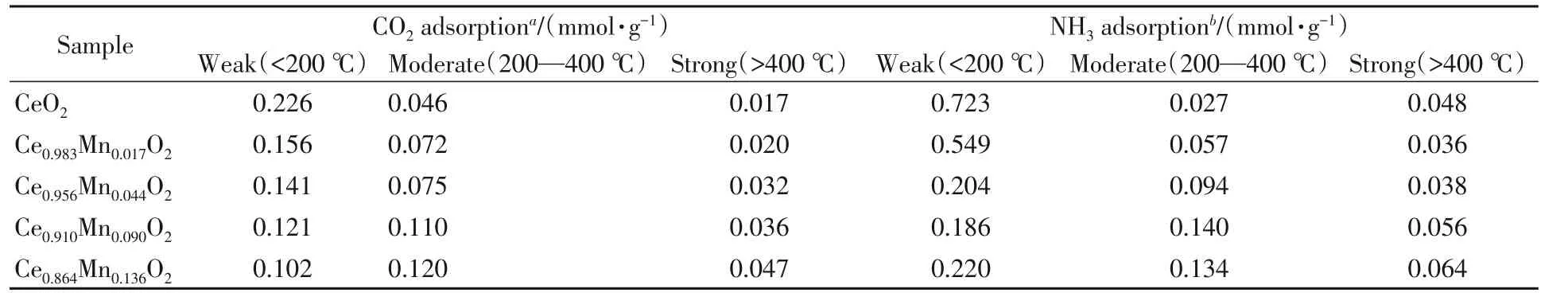
Table 3 Adsorption capacity of CO2 and NH3 of Ce1−xMnxO2 catalysts
图5(A)中,低于200 ℃的脱附峰由CO2在弱碱性吸附位脱附形成,200~400 ℃之间的脱附峰为CO2在中等强度碱性吸附位的脱附,而400 ℃以上的脱附峰为CO2在强碱性位的脱附. 弱碱位的形成主要来源于CO2与催化剂表面羟基结合而生成的碳酸氢盐;中等强度碱性位形成主要来源于CO2与催化剂中的Ce-O对形成的双齿吸附态和桥式吸附态的碳酸盐物种;而强碱性位形成主要来源于CO2与催化剂表面低配位的O2−形成的单齿碳酸盐物种[18]. 由表3可见,催化剂表面弱碱位数量随Mn掺杂量的增加而逐渐降低,而中等强度碱性位和强碱性位数量则随着Mn掺杂量的增加而升高.
图5(B)中,低于200 ℃的脱附峰归属为NH3在弱酸性吸附位的脱附,200~400 ℃之间产生的脱附峰对应于NH3在中强酸性吸附位的脱附,而400~600 ℃之间产生的脱附峰对应于NH3在强酸性吸附位的脱附. 由表3可知,Mn掺杂后催化剂表面酸性位数量的变化与碱性位基本一致. 随着Mn掺杂量增加,催化剂表面弱酸性位数量逐渐减少,中等强度酸性位数量逐渐增加,而强酸性位数量则呈先减少后增加的趋势. 当Mn掺杂量超过1.7%时,随着Mn掺杂量增加,催化剂表面强酸性位数量增加.
2.5 催化剂的表面组成分析
图6(A)为Ce3d的XPS谱图,根据文献[37]报道,Ce3d谱图可分为8个峰,分别为v(约882.4 eV),v′(约885.1 eV),v″(约888.9 eV),v″′(约898.2 eV),u(约900.9 eV),u′(约903.4 eV),u″(约907.4 eV)和u″′(约916.6 eV). 其中,4个u峰和4个v峰分别代表Ce3d3/2和Ce3d5/2自旋轨道分裂峰,u″′,u″,u,v″′,v″和v6 个峰代表Ce4+上3d10f0能级上的电子状态,u′和v′代表Ce3+上3d104f1能级上的电子状态. 采用拟合后的u′和v′峰峰面积之和与u和v峰的总面积的比值来定量计算表面Ce3+的含量,结果列于表4. 另外,由图6(B)可见,催化剂表面同时存在3 种氧物种. 其中,结合能约为529.0 eV 处的峰归属为催化剂表面的晶格氧(OL);结合能约为530.5 eV处的峰归属于催化剂表面的氧空位(OV);结合能约为532.5 eV 处的峰则可归属于催化剂表面的化学吸附氧(OC),包括OH−,CO32−和O-等[38,39].
由表4可知,随着Mn掺杂量增加,催化剂表面Ce3+含量呈现先增加后降低的趋势. 当Mn掺杂量为1.7%时,Ce3+的比例由纯CeO2的10.8%提高为14.6%,这可能是由于前驱体中的Mn 离子价态为+2 价,Mn2+进入CeO2晶格中形成固溶体后发生了Ce4+与Mn2+间的电子传递(Ce4++Mn2+→Ce3++Mn3+)作用,以维持固溶体整体的电中性[19];此外,结合XRD 表征可知,由于Mn 离子半径(Mn2+离子半径为0.083 nm;Mn3+离子半径为0.0645 nm)相对较小,Mnn+取代部分Ce 离子进入CeO2晶体结构中会导致CeO2晶格收缩,该情况出现时,离子半径较小的Ce4+(0.097 nm)会自发向离子半径较大的Ce3+(0.110 nm)转化以弥补晶格的收缩,导致Ce3+比例的提升[40,41]. 然而,Mn掺杂量的进一步增加却导致催化剂表面Ce3+的比例逐渐下降[42,43],这可能是由于催化剂表面Mn 掺杂量及其氧化程度增高使Mn 和CeO2间发生电子作用(Ce3++Mn4+→Ce4++Mn3+),从而造成表面Ce3+比例的减少[19]. 由表4 可见,相比于纯CeO2,Mnn+掺杂后催化剂表面氧空位含量呈先升高后降低的趋势,该变化规律与Ce3+含量变化一致,表明Ce3+含量越高催化剂表面氧空位含量越高[44].
为了进一步验证催化剂表面氧空位变化的原因,对Mn2p的XPS 谱图进行了分析,结果如图7所示. 其中,结合能在650~660 eV 范围内的峰对应于Mn 的2p1/2电子,在较低结合能处的峰由Mn2p3/2电子产生. 根据文献[45,46]报道,Mn2p3/2的XPS 谱图拟合为3 个峰,其中,结合能在约643.4 eV 处的峰归属为Mn4+,而结合能在约641.3 和639.6 eV 处的峰可分别归属为Mn3+和Mn2+. 对其峰面积进行计算后可知催化剂表面不同价态Mn 离子比例(表4). 可以看出,催化剂表面Mn离子存在3种价态且以+3价为主,随着Mn掺杂量增加,表面Mn2+比例逐渐降低,而表面Mn4+比例逐渐升高,与文献[37]报道一致. 原因可能是,当Mn掺杂量增大时,Mn离子易于富集在催化剂表面,在氧化性气氛焙烧的过程中容易被氧化为Mn4+,导致Mn4+比例随Mn掺杂量的增加而升高[47]. 以上结果表明,当Mn 掺杂量较低时,Mn 离子中Mn2+比例较高,有利于Ce4++Mn2+→Ce3++Mn3+反应的进行,进而促进催化剂表面氧空位生成;而当Mn 掺杂量较高时,Mn 离子中Mn4+比例较高,有利于Ce3++Mn4+→Ce4++Mn3+反应的进行,导致催化剂表面氧空位数量减少.

Fig.6 XPS spectra of Ce3d(A)and O1s(B)of Ce1-xMnxO2 catalysts

Table 4 Surface concentrations of Ce,O and Mn estimated by XPS

Fig.7 XPS spectra of Mn2p of Ce1-xMnxO2 catalysts
2.6 催化剂的活性评价与构效关系
催化剂的活性评价结果如图8所示. 可见,随着Mn掺杂量增加,催化剂活性呈先升高后降低的变化趋势. 当Mn 掺杂量为1.7%时,催化剂活性最高,DMC 收率达到11.25 mmol/g,比纯CeO2的活性(9.26 mmol/g)提高了21.49%. 然而,随着Mn掺杂量的进一步增加,催化活性开始降低,当Mn 掺杂量为13.6%时,DMC 收率下降至7.87 mmol/g. 同时,空白实验结果表明,以MnxOy为催化剂,产物中未检测到DMC 生成,说明MnxOy对CO2和甲醇合成DMC 反应不具有催化活性.XRD,BET 和TEM 表征结果显示,Mn 掺杂量并未显著影响催化剂的晶相结构、织构性质和形貌,可见以上因素并不能引起催化活性变化. 研究认为,催化剂表面酸碱性位数量和氧空位含量会显著影响甲醇和CO2合成DMC的反应活性[48].
Ce1−xMnxO2催化剂DMC收率和其表面弱碱/中强碱/强碱及弱酸/中强酸/强酸量变化分别如图9(A)和(B)所示. CO2作为Lewis酸可在中强碱位上形成双齿吸附态,双齿吸附态的CO2与甲醇解离的CH3O*反应生成甲基碳酸酯活性中间体,而CO2的其它吸附方式如碳酸氢盐、单齿吸附态和桥式吸附态则不能完成上述过程[49]. 因此,当Mn 掺杂量较低时,催化剂表面中强碱位数量的增加有利于催化活性的提高. 然而,当Mn掺杂量较高时,伴随着中强碱位数量的增加,强碱位数量也逐渐增加. CO2在强碱性位吸附会形成稳定的CO32−结构,难以与甲醇结合生成活性中间体,造成催化剂活性降低[18]. 催化剂表面的酸性位主要起活化甲醇分子的作用,甲醇分子可在酸性位上解离形成羟基和甲基并吸附在催化剂表面,后者可以与甲基碳酸酯中间体反应生成DMC[50]. 当Mn掺杂量较高时,催化剂表面显著增加的强酸和强碱位还可能会导致生成的DMC发生分解,造成催化活性降低[51].
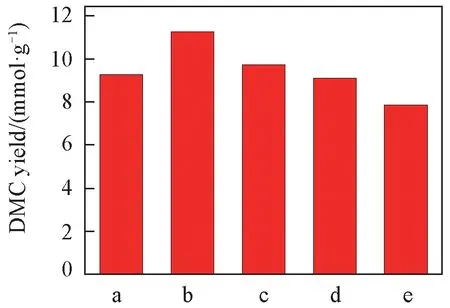
Fig.8 Catalytic activity of Ce1-xMnxO2 catalysts
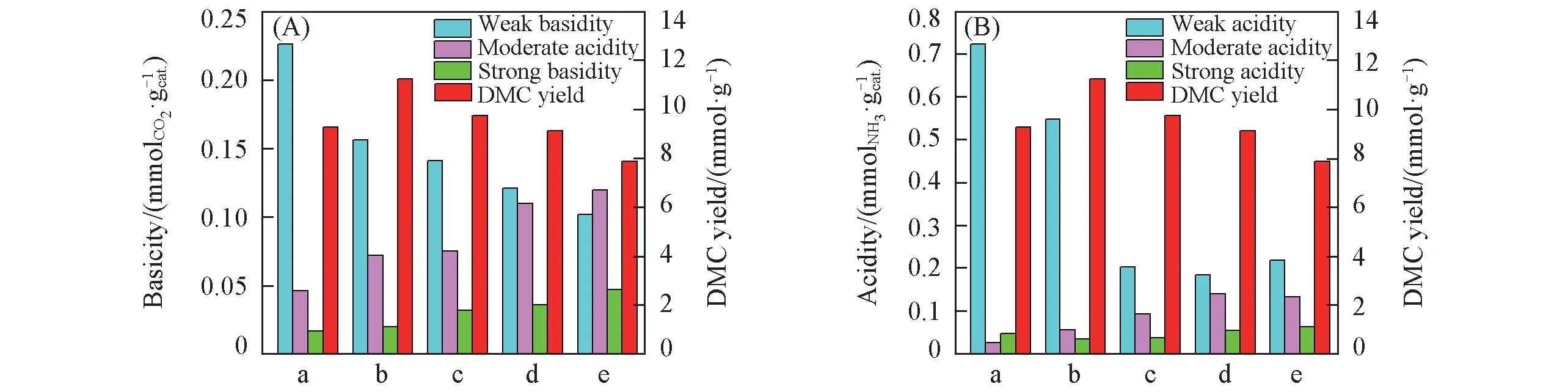
Fig.9 Correlation between catalytic activity and basicity(A),acidity(B)of Ce1-xMnxO2 catalysts
综上所述,中等强度酸/碱量的增加有利于催化活性的提高,而强酸/碱量的增加不利于反应的进行,只有催化剂表面具有适宜的酸碱性才能够有效催化CO2与甲醇反应生成DMC.

Fig.10 Correlation between oxygen vacancies and catalytic activity of Ce1-xMnxO2 catalysts
Ce1−xMnxO2催化剂活性与其表面氧空位含量呈明显线性关系(图10). 研究发现,氧空位作为Lewis碱性位能够促进CO2的吸附、活化和解离[52]. 反应物CO2分子中的一个O 原子可以插入到CeO2表面的一个氧空位当中,使之活化形成双齿吸附态物种,并与甲醇解离产生的CH3O*反应生成甲基碳酸酯中间体,原位红外表征也证实了上述过程[12]. 研究发现,相比于完美晶面,CO2分子在氧空位处的活化具有更低能垒[13]. Mn离子进入CeO2晶格后,由于Mnn+与Ce4+/Ce3+之间的电子效应而引起催化剂表面氧空位含量的变化,进而影响催化剂活性. 氧空位含量的增加有利于增强催化剂对CO2的吸附和活化作用,进而提高催化活性;反之,氧空位含量减少不利于反应物CO2的吸附和活化,催化活性降低.
3 结 论
Ce1−xMnxO2催化剂中Mn 离子可掺杂进入CeO2晶格中形成固溶体. 随着Mn 掺杂量增加,催化剂表面弱酸碱位数量逐渐降低,中强酸碱和强酸碱位数量增加,中等强度酸/碱位数量增加有利于催化活性的提高,而强酸/碱位数量增加不利于反应的进行. 随着Mn掺杂量增加,催化剂表面氧空位含量呈先升高后降低的趋势,这与催化活性变化趋势一致. 当Mn 掺杂量为1.7%时,催化剂的催化活性(11.25 mmol/g)最佳.
猜你喜欢
火炸药学报(2022年5期)2022-11-04 02:30:48
数学物理学报(2019年5期)2019-11-29 07:46:50
物理实验(2019年7期)2019-08-06 05:35:56
航空材料学报(2019年2期)2019-04-15 01:04:08
陶瓷学报(2019年5期)2019-01-12 09:17:38
物理学报(2018年22期)2018-12-18 05:58:28
数学物理学报(2017年5期)2017-11-23 07:51:09
潍坊学院学报(2016年6期)2016-04-18 13:56:55
读者欣赏(2014年6期)2014-07-03 03:00:48
长江大学学报(自科版)(2014年1期)2014-03-20 13:20:12
