
小鼠诺如病毒数字RT-PCR 定量检测方法的建立
2020-08-17 01:00谭建锡禹思宇胡忆文周智君唐连飞
湖南畜牧兽医 2020年3期
谭建锡,禹思宇,胡忆文,周智君,唐连飞※
(1.长沙海关技术中心,湖南长沙410004;2.中南大学实验动物学部,湖南长沙410008)
小鼠诺如病毒(murine norovirus, MNV)是Stephanie M.Karst 等于2003 年在基因缺失小鼠中分离到的[1]。几乎所有的实验小鼠对此病毒易感,在美国、加拿大、日本等国的实验小鼠体内均监测到该病毒[2],国内小鼠中的感染率也比较高[3]。MNV的诊断方法目前主要为分子生物学方法[4-6]和血清学方法e。血清学方法由于受到抗原数量少,检测耗时长等因素影响,其实际应用受到限制。荧光定量RT-PCR 法等分子方法因具有较好的灵敏度和特异性,应用更为广泛,但其定量测定需要依赖标准品建立标准曲线来进行,操作耗时费力。
微滴数字PCR(droplet digital PCR,ddPCR)是一种用于核酸绝对定量的新兴技术,可直接获得样本中极低含量基因片段拷贝数。由于基于统计学方法和泊松(poisson)分布,可不需标准曲线就可实现对低浓度核酸的高灵敏度绝对定量,使得ddPCR 技术广泛的应用于核酸绝对定量领域。已有将该技术应用于H7N9 流感病毒[8]、食源性致病菌[9]和伪狂犬病毒[10]定量研究的报道,弥补了常规检测的不足。
本研究将微滴数字PCR 技术应用于MNV 检测,并对其最佳退火温度、检出限以及检测准确度等方面进行研究,建立了MNV 的dd RT-PCR 绝对定量方法,为MNV 的定量检测提供了技术支撑。
1 材料与方法
1.1 试验材料
MNV 核酸、小鼠肺炎病毒核酸、呼肠孤病毒3 型核酸均由长沙海关技术中心动物检疫实验室保存。
1.2 试剂和仪器
QX200 微滴式数字PCR 仪、微滴发生卡、PX1热封仪及卡垫(美国Bio-Rad 公司);LC480II 荧光PCR 仪(瑞士Roche 公司);LabCycler 96 梯度PCR扩增仪(德国LabCycler 公司)。
ddPCR droplet 生 成 油、One-step RT-ddPCR Advanced 试剂盒、Droplet Reader 油:(美国Bio-Rad公司);qRT-PCR 一步法试剂(EvoScript RNA Probes Master)(瑞 士Roche 公 司);QIAamp Viral RNA Mini 试剂盒(QIAGEN 公司);
1.3 引物
MNV 微滴数字RT-PCR 引物和探针以ORF1、ORF2 高度保守的结合区为靶向扩增区域[11](见表1),引物及探针由上海生工生物工程公司合成。
1.4 RNA 提取
按照QIAamp Viral RNA Mini Kit 说明书,对MNV 基因组RNA 进行提取,提取好的RNA 保存于-80 ℃冰箱中备用。
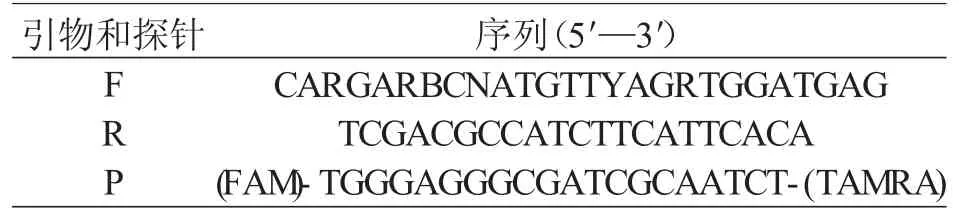
表1 ddRT-PCR 引物和探针序列
1.5 dd RT- PCR 反应方法构建
反应体系(20 μL)如下:5 μL 4×Supermix,浓度均为900 nmol/L 的上、下游引物终,终浓度为250 nmol/L 探针,以及1 μL 300 nmol/L DTT、2 μL RNA 模板,每个反应用ddH2O 补足至20 μL。生成微滴操作:将微滴生成油70 μL 和反应液20 μL,分别加入到最后一排的各孔和DG8 微滴生成卡中间位置,覆盖专用胶垫后,将卡片放置于微滴生成仪上,在第1 排中自动生成微滴后,吸取40 μL 加入96 孔板中。扩增:在PX1 热封仪上放置96 孔板,设置封膜程序180℃10 s,放入梯度PCR 扩增仪进行扩增。反应条件为:50℃60 min,95℃10 min;95℃15s,最佳退火温度1min,40 个循环,98℃10 min。数据分析:扩增后在微滴式数字PCR 仪QX200 中进行信号读取,获取并分析试验数据。用水作为阴性对照模板。
1.6 退火温度条件优化
设计退火范围梯度为55.5℃~62.5℃,温度间隔为1℃,共设置8 个梯度。按照上述1.5 中的步骤,扩增时在PCR 仪上设置相应扩增温度,扩增后对数据进行分析;用核酸进行试验,重复进行3 次,求平均值,确定最佳退火温度。
1.7 灵敏性及重复性试验
按10 倍梯度将病毒核酸进行稀释,取2 μL 作为模板,按照已建立的ddRT-PCR 方法,绘制线性标准曲线并测定最低检出限。并按照“1.5”和“1.6”中确定的最佳条件,进行反应体系和反应条件设置。进行3 次重复操作,计算变异系数,评估重复性。
1.8 特异性试验
以MNV 核酸、小鼠肺炎病毒核酸、呼肠孤病毒3 型核酸为模板进行特异性实验,检测方法的特异性,以水做阴性对照。
1.9 临床应用
随机取20 个小鼠盲肠样本进行MNV 检测。将小鼠盲肠样本,制备匀浆后提取RNA,以qRT-PCR和上述建立的ddRT-PCR 方法进行检测,对比检测结果的一致性。
2 结果与分析
2.1 ddRT- PCR 方法建立
依据试验指南及相关文献,对反应体系进行制备,优化退火温度,建立了ddRT-PCR 检测方法。如图1 显示,在55.5℃~62.5℃退火范围内均检出荧光信号,但随着温度升高扩增振幅有降低趋势。在55.5℃~58.5 ℃时出现较高的扩增振幅,且阴阳性之间存在较为明显的微滴分界,但两者之间也略微存在微滴弥散的现象。分析由于MNV 的基因型比较复杂,检测引物和探针的设计中常常需要简并碱基,较低的退火温度降低了产物扩增特异性,因此在ddRT-PCR 阳性微滴和阴性微滴之间出现弥散现象。综合对扩增振幅和产物扩增特异性考虑,选择57.5℃作为反应最佳退火温度。
从图2 中可以看到,随着由左到右(D04—D11)扩增温度依次升高,反应体系中可检测到的阳性微滴数量有逐渐减少的趋势,这也与图1 中显示的扩增振幅随着温度升高振幅降低的变化趋势一致;直方图显示(图3),阴性微滴峰荧光强度值较低而阳性微滴峰的荧光强度值普遍较高,且两者之间具有较明显的区分,不存在其他杂峰,易于阴阳性结果的判定。以上结果说明,建立的ddRT-PCR方法具有较高的可靠性。
2.2 标准曲线和最低检测限
根据梯度浓度稀释法来测定检测方法的灵敏度。以核酸稀释度和阳性拷贝数的对数值绘制标准曲线(图4),发现线性方程为y =-0.8796x +5.0315,线性关系R2值为0.985,最低检测限为7 copies/μL,表明建立的ddRT-PCR 方法具有较好的较低的检测低限及线性灵敏度。
2.3 ddRT- PCR 重复性分析
利用建立的ddRT-PCR 方法,对同一核酸模板进行3 次重复试验,发现变异系数均<7%,证明所建立的ddRT-PCR 检测方法重复性良好,检测结果稳定、可靠(表2)。

表2 ddRT-PCR 重复性试验结果
2.4 ddRT- PCR 特异性试验
以MNV 核酸、小鼠肺炎病毒核酸、呼肠孤病毒3 型核酸为模板进行ddRT-PCR 扩增后,观察各孔微滴数,生成量均衡,试验成立。由图5 可知,仅有MNV 核酸孔(D04)检测到阳性微滴,小鼠肺炎病毒核酸孔(D05)、呼肠孤病毒3 型核酸孔(D06)及阴性对照孔(D07)中均未出现阳性微滴信号,表明该检测方法检测诺如病毒与其他病毒均不存在交叉反应,具有良好的特异性。
2.5 临床样品验证
将20 份小鼠盲肠样品提取RNA 后,分别用qRT-PCR 方法和已建立的ddRT-PCR 方法进行检测,检测结果发现1 份样品为阳性,其余19 份样品均为阴性。两种方法的检测结果一致,表明已建立的ddRT-PCR 方法有较好的实用性。
3 讨论
MNV 给人类的健康带来了严重威胁,感染病毒后会出现剧烈腹泻、呕吐等一系列相关症状,严重感染者还可能导致心律失常、性肾功能衰竭等[12]。传统的MNV 检测方法主要是电镜法(EM)、放射性免疫试验(RIA)、酶联免疫法(ELISA)。但诺如病毒在电镜下无显著形态学特征,难以满足实际需求;RIA法实验时间需长,且要标记放射性同位素,影响操作环境;ELISA 由于抗原数量少,其实际应用受到限制,灵敏度较低,目前已逐渐被RT-PCR 和实时荧光RT-PCR 技术取代[13]。随着技术的不断进步,荧光RT-PCR 方法在病毒定性检测中应用较多,但其定量测定过程中,需要对样品初始拷贝数利用添加外标来定量,操作步骤耗时费力。利用数字微滴PCR 技术对病毒进行检测时,摆脱了对标准品的依赖,不需要建立标准曲线就可进行绝对定量,最低可检测到单个拷贝;且对反应条件要求较低,受基质中抑制剂的影响较小等[14]优点,使其在检测低含量病毒和病毒的定量测定方面相比荧光RT-PCR方法有较大优势。
本研究通过微滴数字PCR 技术,建立了MNV 的ddRT-PCR 绝对定量方法,确定了最佳的退火温度及最低检出限,实现了从样品中直接获得病毒核酸拷贝数的绝对定量和高灵敏度检测,为MNV 定量检测提供了一种新思路。其不足之处在于,利用微滴生成卡单次产生微滴量较低,在检测大量样品时需进行重复操作,因此在高通量检测方面仍需改进。随着未来技术进步,高质量引物设计软件的出现及微流控高通量微滴生成卡片的研制,将会不断提升ddPCR 技术的检测效率,使其应用于更广泛的领域中。
猜你喜欢
中国慈善家(2022年3期)2022-06-14
中国农学通报(2022年12期)2022-06-01
现代苏州(2022年9期)2022-05-26
中学生数理化·高一版(2022年4期)2022-05-09
中国糖料(2022年2期)2022-04-06
快乐语文(2021年34期)2022-01-18
中国种业(2021年11期)2021-11-25
食品安全导刊(2021年21期)2021-08-30
中国(俄文)(2020年8期)2020-11-23
中学生物学(2019年7期)2019-10-17
