
基于CRISPR/Cas9技术构建人CLCN7基因编辑载体
2020-06-29 07:43孙国平马驰宇朱鹏薛雯龚蔚蔚欧明林
实验与检验医学 2020年3期
孙国平 ,马驰宇 ,朱鹏 ,薛雯 ,龚蔚蔚 ,欧明林 ,4
(1.深圳市坪山区人民医院中心实验室,广东 深圳518118;2.广西师范大学生命科学学院,广西 桂林541004;3.中国人民解放军第九二四医院中心实验室,广西代谢性疾病研究重点实验室,广西 桂林541002;4.深圳市人民医院临床医学研究中心,广东 深圳 518020)
石骨症是一种骨代谢异常疾病,也是一种典型的人类遗传病。近年来,DNA 测序技术的发展促进了人类遗传病致病突变点的发现[1]。目前,人类已发现数十个与常染色体显性遗传石骨症密切相关的CLCN7 突变位点,其中CLCN7(R286W)突变是最常见的致病突变位点之一,可见于40%以上的石骨症患者[2]。该病尚缺乏有效的治疗方法,参考遗传病研究领域最新研究进展,探索石骨症致病基因点突变修复方法具有重要的科学价值,但尚缺乏相关报道。因此,办研究针对较常见的致病突变点构建基因编辑载体,可能为石骨症致病基因CLCN7修复提供研究基础。
1 材料与方法
1.1 材料
1.1.1 质粒与菌株 CRISPR/Cas9 质粒购自北京赛贝生物技术有限公司,质粒图谱见图1。感受态细胞DH5α 购自天根生化科技(北京)有限公司。
1.1.2 试剂 BspQ I 高保真限制性内切酶和T4 连接酶购自NEB 公司;DNA 凝胶回收试剂盒和质粒小提试剂盒及TOP10 感受态细胞购自天根生化科技(北京)有限公司;高保真PCR 酶购自宝日医生物科技(北京)有限公司。
1.1.3 引物合成与测序寡链核苷酸(oligo) DNA 引物由苏州金唯智生物科技有限公司提供,DNA 序列测序由北京六合华大基因科技有限公司完成。
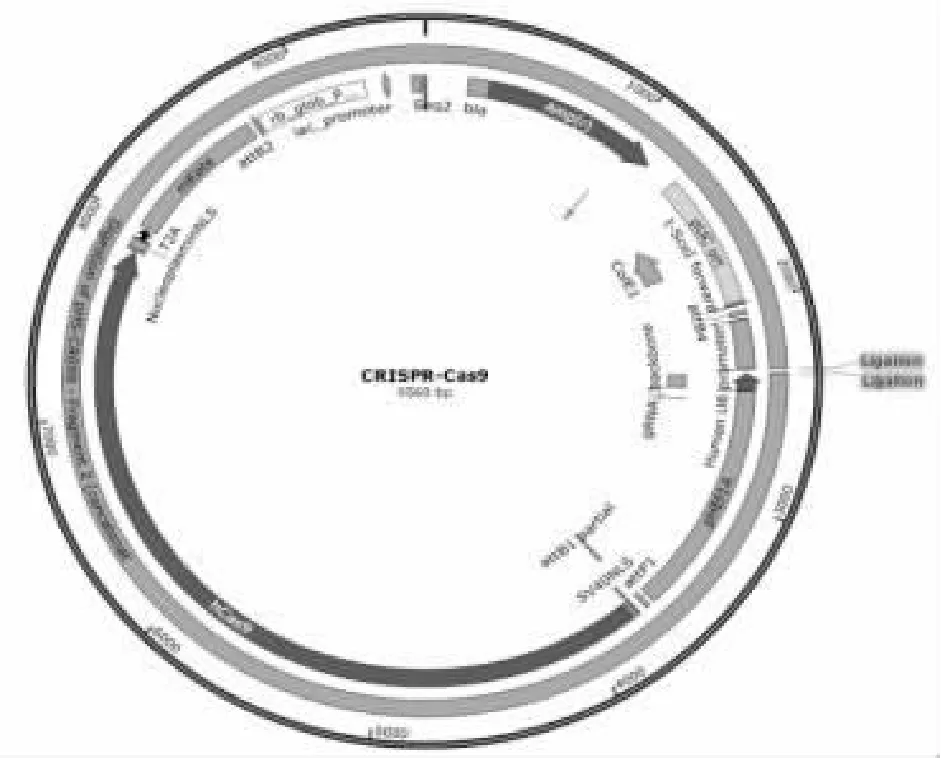
图1 CRISPR /Cas9 质粒图谱
1.2 方法
1.2.1 sgRNA oligo 序列的设计 根据 https://www.nlm.nih.gov/网站确认 CLCN7 基因序列(S1);根据http://crispr.mit.edu/网站设 计 在 CLCN7 基 因 序 列设计sgRNA 序列。其中sgRNA 的设计原则为:起始为碱基为G,靶位点后序列的PAM 序列为NGG;Forward 链 5` 端添加 CACC,Reverse 链的 5` 端添加AAAC,并经过BspQ I 酶切后形成的粘性末端进行互补形成闭环质粒。

表1 CLCN7-sgRNA 序列和基因组检测引物序列
1.2.2 重组质粒CLCN7-sg1RNA-CRISPR/Cas9 和CLCN7-sg2RNA-CRISPR/Cas9 的构建 利用 Bsp QI 对 CRISPR/Cas9 质粒进行酶切,通过 Agarose 凝胶电泳分离目的条带后,由胶回收方法获得线性化载体;对sg1RNA 和sg2RNA 进行退火实验后,分别使用T4 DNA 连接酶将退火后的产物连接至线性CRISPR/Cas9 载体,实验反应条件:16℃,过夜。将连接产物转化至TOP10 感受态细胞中, 涂布在含有氨苄青霉素抗性的LB 平板上,进行克隆筛选,并使用PCR 的方法(表1)鉴定目片段是否已插入,最后挑取阳性克隆进行Sanger 测序,确认插入片段的序列。
2 结果与分析
2.1 sg1RNA 和sg2RNA 的退火 将合成的sg1RN A-F / sg1RNA-R 和 sg2RNA-F / sg2RNA-R 序列,稀释至10 μm 后,两对引物以1:1 比例进行混合,在95 ℃下孵育 10 min, 移至室温下自然冷却2 h以上,琼脂糖凝胶电泳分析发现双链寡核苷酸片段sg1RNA 和sg2RNA 电泳条带与目的片段大小一致(图2)。
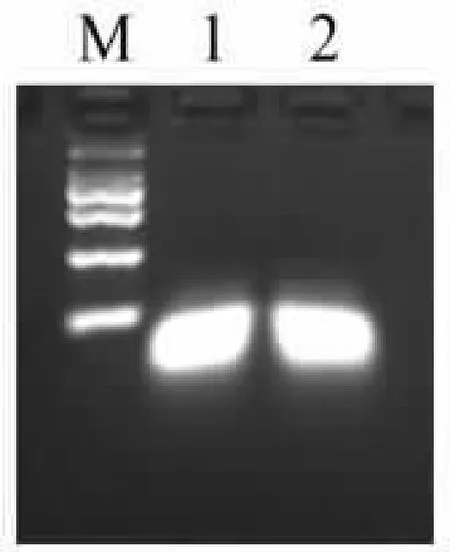
图2 双链的寡核苷酸片段sg1RNA 和 sg2RNA 的检测
2.2 CRISPR/Cas9 质粒酶切 质粒 CRISPR/Cas9 序列上具有BspQ I 酶切位点,见图1;采用BspQ I 酶切断质粒并且去磷酸化后,琼脂糖电泳检测发现酶切获得的DNA 片段为9.5 kbp,与预期大小一致,见图3。
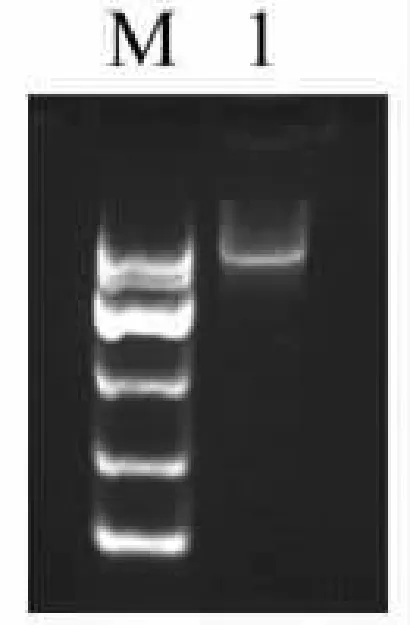
图3 CRISPR/Cas9 质粒的BspQ I 限制性内切酶的酶切鉴定
2.3 重组质粒 CLCN7-sg1RNA- CRISPR/Cas9 和CLCN7-sg2RNA-CRISPR/Cas9 构建 在重组质粒CLCN7-sg1RNA-CRISPR/Cas9 和CLCN7-sg2RNA-CRISPR/Cas9 的构建过程中, 分别将线性化载体与寡核苷酸使用T4 连接酶连接, 并转化至TOP10细胞中,并分别挑取8 个克隆,扩培后提取质粒,使用 DNA 引物 CC-CX-F 和 CC-CX-R 进行 PCR分析,见表1 与图4,结果表明除了重组质粒CLCN7-sg1RNA- CRISPR/Cas9 的 6 号克隆和 CLCN7-sg2RNA-CRISPR/Cas9 的16 号克隆外均为阳性克隆;最后,选取阳性克隆进行Sanger 测序,结果显示阳性克隆成果插入了目的序列sg1RNA 和sg2R NA,碱基序列与预期一致,见图5 和图6,证实重组质粒 CLCN7-sg1RNA- CRISPR/Cas9 和 CLCN7-sg2RNA-CRISPR/Cas9 已构建成功。
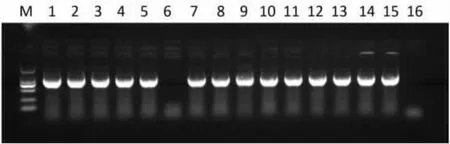
图4 PCR 鉴定重组质粒

图5 重组质粒CLCN7-sg1RNA- CRISPR/Cas9 的测序结果
3 结论与讨论
CRISPR/Cas 系统存在于大部分细菌和古细菌中,最早由日本大阪大学院研究员发现[3]。近年来,不少研究证实CRISPR/Cas 是细菌利用无义RNAs标记外源核酸序列引导Cas 蛋白实现特异性切割的防御系统[4]。Cas9 是Cas 蛋白的一种,是Ⅱ型CR ISPR 系统的核心蛋白,CRISPR 基因座转录形成的crRNA,经 tracrRNA (transactivating chimeric RNA)促进成熟后,可与tracrRNA 结合共同介导Cas9 特异性切割目标DNA[5]。目前,研究证实 crRNA 和tracrRNA 可以进行融合,作为sgRNA,可引导Cas9蛋白定点敲除或编辑目的基因,CRISPR/Cas9 已成为了一种新型的基因编辑工具[6,7]。
本文基于CRISPR/Cas9 技术尝试构建了CLCN7 基因编辑表达载体。研究表明,为了实现对靶基因的编辑,首先要根据靶基因的识别序列设计特异性sgRNA,但是值得注意的是sgRNA 在识别靶位点时容易出现碱基错配或者形成DNA/RNA凸起,导致CRISPR/Cas9 系统产生脱靶效应[8]。Cas9介导的DNA 双链断裂产生在PAM 区上游的3bp处,靠近PAM 的7~12bp 序列为决定其特异性的关键[9]。因此,在设计sgRNA 时要预测其潜在脱靶位点,减少sgRNA 与脱靶位点的碱基配对数,且在靠近PAM 区至少有2 个碱基不配对,避免有连续或间隔的4 个碱基配对[10,11],该策略可以提高sgRNA特异性。遵循上述原则,我们在CLCN7 外显子区设计了两条长20bp 的sgRNA,其靶点序列的间隔为3bp,并在其后端紧邻PAM(前间区序列邻近基序,NGG)[12,13]。其与靶位点的结合有待后续的实验证实。
本研究选用含U6 启动子和Cas9n 蛋白酶的CRISPR-Cas9 质粒,经BspQI 酶切得到线性载体,使用T4 连接酶将线性载体与两对sgRNA 退火后得到的寡核苷酸链分别连接,经鉴定,成功构建了CLCN-7 基因编辑表达载体 (CLCN7-sg1RNACRISPR-Cas9 和 CLCN7-sg2RNA-CRISPR-Cas9),本实验构建载体的成功率较高,为下一步修复CLC N7 致病点突变提供了可能。
据现有文献报道,石骨症的有效治疗手段较为有限,其中同种异体造血干细胞移植是治疗严重石骨症的有效方法之一, 但该方法受到多种因素限制,未能在良性石骨症治疗中得到应用[14]。最近,一些研究表明通过患者自身体细胞建立诱导多能干细胞,并进行致病突变修复和定向分化,可能应用于石骨症治疗[15]。因此,基于CRISPR/Cas9 技术探索构建人CLCN7 基因编辑载体具有积极的意义,可能为下一步石骨症特异诱导多能干细胞CLCN7突变修复提供新的信息和基础。
猜你喜欢
环球时报(2022-09-20)2022-09-20
——一道江苏高考题的奥秘解读和拓展
中学生物学(2022年7期)2022-09-07
成都医学院学报(2022年4期)2022-08-19
中国农学通报(2022年12期)2022-06-01
中国糖料(2022年2期)2022-04-06
中国种业(2021年11期)2021-11-25
教学考试(高考生物)(2021年2期)2021-05-31
江西农业学报(2021年4期)2021-04-20
今日农业(2020年24期)2020-12-15
三农资讯半月报(2020年11期)2020-06-21
