
利用CRISPR/Cas9技术敲除转基因水稻中的潮霉素标记基因
2020-06-22 08:36彭梅芳潘春梅宋健兰范晓丽陈克贵
西南农业学报 2020年5期
彭梅芳,甘 凤,潘春梅,宋健兰,范晓丽,陈克贵
(四川省农业科学院生物技术核技术研究所,四川 成都 610061)
【研究意义】在植物基因工程中,通常将目标基因(Gene of Interest, GOI)表达元件与适宜的选择标记基因(Selectable Marker Gene, SMG)构建到同一载体上,一起转入植物细胞,然后借助SMG筛选转化细胞,进一步培育转基因植株[1]。通过这种方法得到的转基因植株中SMG会随着GOI一起整合到宿主植物基因组中,而常用的SMG编码的蛋白通常是抗生素或除草剂类,SMG残留可能对人畜等食物链下游生物产生不良影响,特别是对人体健康存在潜在危害,这也是转基因生物安全性一直备受争议的主要原因[2];此外,目前可利用的SMG数量有限,利用相同的SMG,对植物多次导入GOI存在一定困难,阻碍了多次转化达到基因聚合的目的[3]。并且我国转基因生物安全管理也要求无标记基因的存在。因此,无论从转基因生物安全性考虑,还是从基因工程技术的更广泛应用考虑,去除转基因作物中的SMG都十分必要。 【前人研究进展】基因组编辑是指利用人工序列特异核酸酶(Sequence-specific nucleases, SSNs)在基因组特定位点产生DNA双链断裂(Double-strand breaks, DSBs),实现对基因靶位点的敲除、染色体的重组以及基因的定点插入或替换等,是对基因组进行定向修饰的一种技术。根据所采用的人工核酸内切酶不同,基因组编辑技术主要经历了三代发展:即锌指核酸酶(Zinc finger nucleases, ZFNs)[4]技术、类转录激活子样效应因子核酸酶(Transcription activator-like effector nucleases, TALENs)[5]技术、成簇的、规律间隔的短回文重复序列(clustered regularly interspaced short palindromic repeats, CRISPR)及其相关核酸酶9系统(CRISPR-associated 9, Cas9)[6]。CRISPR/Cas9系统自2013年初建立以来[7-8],因其具有操作简单、作用高效、成本低廉等优点,迅速成为基因组改造与修饰的重要工具。目前已成功应用于各种细菌、动物及植物的基因组精确修饰中,被认为是最具有广阔应用前景的一种基因组定点编辑技术。【本研究切入点】本研究利用CRISPR/Cas9基因编辑技术,针对水稻转基因中常用的SMG潮霉素抗性基因(hygromycinphosphotransferaseII,hptII)进行基因编辑。【拟解决的关键问题】期望获得删除hptII基因的转基因水稻,获得marker-free的转基因水稻。
1 材料与方法
1.1 植物材料
受体材料为野生型水稻(OryzasativaL.)品种日本晴(Nipponbare),由四川省农业科学院生物技术核技术研究所基因与基因技术实验室保存。含hptII基因的转基因水稻品种日本晴由四川省农业科学院生物技术核技术研究所基因与基因技术实验室获得(转化质粒为pCXUN)。
1.2 菌株与质粒
农杆菌LBA4404由四川省农业科学院生物技术核技术研究所基因与基因技术实验室保存。质粒pYLsgRNA-OsU6a(GenBank登录号:KR029105)、pYLsgRNA-OsU3(GenBank登录号:KR029103)和质粒pYLCRISPR/Cas9Pubi-B由华南农业大学刘耀光教授惠赠。质粒pCXUN(GenBank登录号:FJ905215)由中国农业科学院植物保护研究所王国梁研究员惠赠。
1.3 主要酶及试剂
质粒提取试剂盒、胶回收试剂盒和RNA提取试剂盒均购自于天根生化科技有限公司;PrimeSTAR max DNA Polymerase、反转录试剂盒、pMD19-T载体购自TakaRa公司;普通PCRmix购自南京诺唯赞生物科技有限公司;BsaI内切酶购自于New England Biolabs(NEB)公司;T4DNA 连接酶购自MBI Fermentas公司;CTAB、氯仿、异戊醇、异丙醇等常规化学试剂购自上海生工生物工程有限公司;PCR产物测序和引物合成由成都擎科梓熙生物技术有限公司完成。
1.4 CRISPR/Cas9编辑载体构建
根据sgRNA的设计原理,以pCXUN载体中的hptII基因序列为目标序列,通过在线网站CRISPR RGEN Tools(http://www.rgenome.net/cas-designer/)设计hptII基因的sgRNA,选择2个sgRNA作为基因编辑位点,具体sgRNA设计和载体构建主要参照华南农业大学刘耀光教授的方法[9]。
1.4.1 OsU6a+sgRNA-1表达盒的克隆 sgRNA-1目标序列为CTCCGACCTGATGCAGCTCTCGG。第一轮PCR扩增:以U-F+OsU6aHy-1,gRHy-1+gR-R分别各为一对引物(表1),以pYLgRNA-OsU6a质粒为模板,用高保真酶PrimeSTAR进行PCR扩增,分别获得片段1和2。第二轮PCR扩增:以第一轮扩增回收的1和2产物混合作为模板,以Pps-GGL和Pps-GG2-1为引物(表1),PCR获得带BsaI酶切位点的DNA片段,该片段经BsaI酶切后得第一个表达盒OsU6a+sgRNA-1。
1.4.2 OsU3+sgRNA-2表达盒的克隆 sgRNA-2目标序列为ACAGACGTCGCGGTGAGTTCAGG。第一轮PCR扩增:以U-F+OsU3Hy-2,gRHy-2+gR-R分别各为一对引物(表1),以pYLgRNA-OsU3质粒为模板,用高保真酶PrimeSTAR进行PCR扩增,分别获得片段3和4。第二轮PCR扩增:以第一轮扩增回收的3和4产物混合作为模板,以Pps-GG2-2和Pgs-GGR为引物(表1),扩增获得带BsaI酶切位点的DNA片段。该片段经BsaI酶切后得到第二个表达盒OsU3+sgRNA-2。
1.4.3 sgRNA表达盒连入编辑载体 质粒pYLCRISPR/Cas9Pubi-B经BsaI酶切回收载体大片段,与经BsaI酶切后的2个表达盒OsU6a+gRNA-1、OsU3+gRNA-2进行连接,最终获得编辑hptII基因的载体质粒,命名为PMF117。将PMF117转入农杆菌LBA4404中,用于水稻遗传转化。

表1 本研究所用引物
注:ggtctc为BsaI识别位点;ACTAGT为SpeI酶切位点;ACGCGT为MluI酶切位点。
Notes: ggtctc is theBsaI digestion recognition site; ACTAGT is theSpeI digestion recognition site; ACGCGT is theMluI digestion recognition site.
1.5 农杆菌介导的水稻遗传转化
日本晴遗传转化步骤参照Nishimura等[10]的方法,具体地将野生型水稻日本晴种子经75 %无水乙醇、0.1 %升汞消毒处理后诱导成愈伤组织,然后用农杆菌去侵染愈伤组织,经筛选、分化等步骤得到再生植株。
1.6 T0代转基因阳性苗鉴定及cas9表达分析
经双丙胺膦筛选得到的抗性植株,即为T0代转化植株。通过CTAB法提取T0代植株基因组DNA,分别用引物F-Barr+R-Barr和SP-L1+SP-R(表1)进行PCR扩增,2对引物均检测为阳性的植株,即为所需要的转基因阳性植株。进一步对阳性植株提取RNA,反转录成cDNA,用引物F-Lcas9-RT+R-Lcas9-RT(表1)进行RT-PCR检测Cas9基因表达情况。Cas9基因表达阳性的植株经两代筛选获得可用于编辑hptII基因的纯合株系。
1.7 敲除转基因水稻中的hptII基因
将获得的编辑hptII基因的水稻与含有hptII基因的转基因水稻进行杂交,杂交F1代种子萌发,分单株提取幼叶基因组DNA,用潮霉素抗性基因特异引物F-HPTII-542和启动子引物R-35S-P(表1)进行PCR扩增,PCR产物送测序,测序为双峰的即是hptII基因有被编辑的植株。进一步将测序为双峰的PCR产物与T载体pMD19-T连接,进行亚克隆,挑取单克隆进行测序分析hptII基因ORF区序列情况,确定hptII基因是否被敲除。
2 结果与分析
2.1 sgRNA靶点设计和表达盒扩增
以pCXUN载体(GenBank登录号:FJ905215)中的hptII基因序列设计了2个sgRNA的靶位点。sgRNA-1:5’-CTCCGACCTGATGCAGCTCTCGG-3’(正向);sgRNA-2:5’-ACAGACGTCGCGGTGAGTTCAGG-3’(反向)。其中sgRNA-1用OsU6a作为启动子,经重叠PCR扩增得到OsU6a+sgRNA-1表达盒的片段长度为629 bp;sgRNA-2用OsU3作为启动子,PCR扩增得到OsU3+sgRNA-2表达盒的片段长度为603 bp(图1)。
2.2 CRISPR/Cas9基因编辑工程菌的获得
将OsU6a+sgRNA-1和OsU3+sgRNA-2表达盒片段回收后,与质粒pYLCRISPR/Cas9Pubi-B(华南农业大学刘耀光教授惠赠,GenBank登录号:KR029110)一起用BsaI 和T4DNA 连接酶进行酶切连接反应,转化大肠杆菌DH5а,经PCR鉴定阳性克隆送测序,结果表明OsU6a+sgRNA-1和OsU3+sgRNA-2表达盒均准确连入载体pYLCRISPR/Cas9Pubi-B中,命名为PMF117(图2)。将PMF117转化农杆菌LBA4404即获得编辑hptII基因的工程菌。
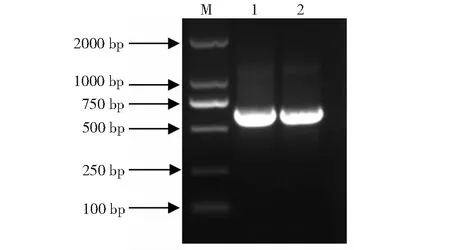
M:DNA marker (DL2000); 1:OsU6a+sgRNA-1表达盒;2:OsU3+sgRNA-2表达盒M: DNA marker (DL2000); 1: OsU6a+sgRNA-1 expression box; 2: OsU3+sgRNA-2 expression box图1 sgRNA 表达盒PCR扩增图Fig.1 PCR amplification of sgRNA expression box
2.3 水稻T0代转基因植株的获得及鉴定
采用农杆菌介导的方法,将PMF117转化到水稻品种日本晴中,得到25株T0代转化植株,分别提取基因组DNA,进行PCR鉴定,其中23株Bar基因和sgRNA表达盒均检测为阳性(图3),阳性率达92 %。进一步对23株阳性植株提取RNA,反转录成cDNA,检测Cas9基因表达情况,结果表明有18株转基因植株中Cas9基因表达为阳性(图4),证明PMF117成功转入水稻植株中,并且Cas9基因得到了表达。

1~6:不同的T0代转基因株系;N:野生型日本晴1-6: Transgenic T0 plantlets; N: Wild type Nipponbare图4 不同转基因植株的Cas9基因转录水平的RT-PCR分析Fig.4 RT-PCR analysis of Cas9 gene transcript in the transgenic plantlets
2.4 hptII基因编辑材料的获得及鉴定
T0代转基因阳性植株再经两代筛选获得纯合的、稳定表达Cas9基因和sgRNA的转基因植株,即获得可以编辑hptII基因的转基因纯合植株。将得到的编辑hptII基因的水稻植株与含有hptII基因的水稻植株进行杂交,CRISPR/Cas9系统即可在杂交F1代植株中对hptII基因的DNA进行编辑剪切。用引物F-HPTII-542和R-35S-P对F1代植株DNA进行hptII基因扩增,PCR产物直接测序分析。结果表明,9株F1代植株中有6株的PCR产物测序在设计的sgRNA位点附近出现明显双峰(图5),初步确认这6株F1代植株的hptII基因有被编辑,编辑率为66.67 %。进一步,将测序为双峰的PCR产物与T载体连接,进行亚克隆,挑取单克隆进行测序,结果表明在F1代植株中hptII基因的ORF区有碱基缺失(1~43 bp)、替换以及单碱基插入(图6),使得hptII基因不能成功翻译。这些编辑的F1代植株通过自交得到F2代,在F2代中筛选可得到hptII基因DNA缺失、不能翻译出HPTII抗性蛋白,并且不含编辑载体等外源片段的植株,即得到成功敲除转基因水稻中的潮霉素标记基因的植株。
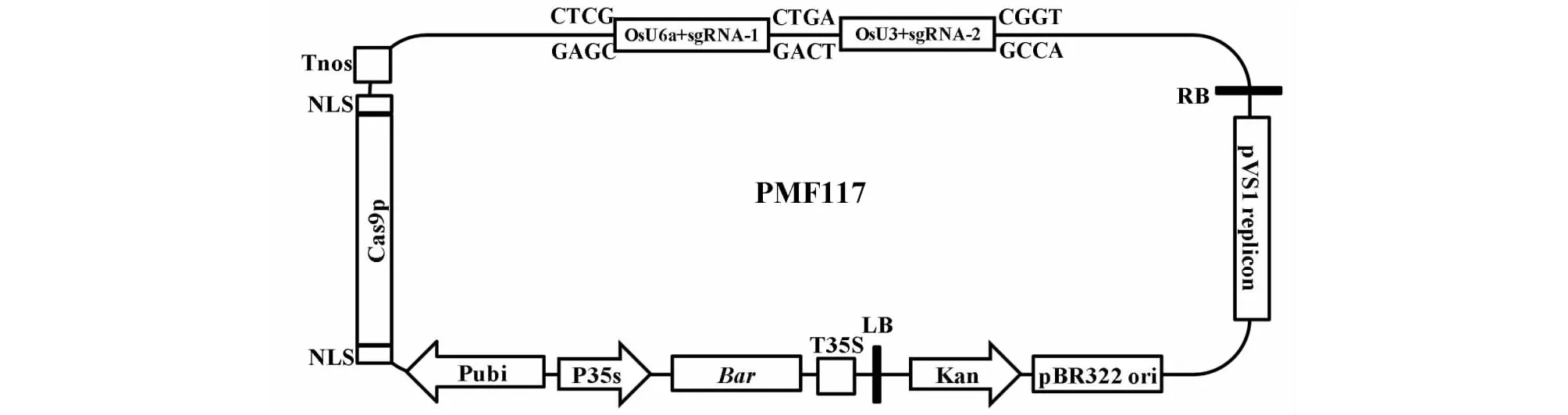
图2 PMF117质粒Fig.2 PMF117 vector
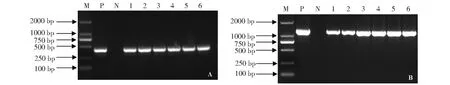
A:转基因植株的Bar基因PCR检测;B:转基因植株的sgRNA表达盒PCR检测;M:DNA marker (DL2000);P:PMF117质粒;N:野生型日本晴DNA;1~6:不同的T0代转基因株系A: PCR detection of Bar gene; B: PCR detection of sgRNA expression box; M: DNA marker (DL2000); P: PMF117 plasmid; N: Wild type Nipponbare DNA; 1-6: Transgenic T0 plantlets图3 转基因植株的PCR检测Fig.3 PCR detection of transgenic plantlets
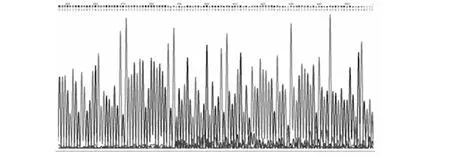
图5 F1代植株的PCR产物测序结果Fig.5 Sequencing results of PCR products from F1 generation plants

A:sgRNA-1靶位点的突变序列比对;B:sgRNA-2靶位点的突变序列比对;G、J、H:不同的F1代植株A: DNA sequence alignment of sgRNA-1 target site; B: DNA sequence alignment of sgRNA-2 target site; G,J,H: Different F1 generation plants图6 亚克隆中 hptII基因靶位点突变序列比对Fig.6 DNA sequence alignment of hptII gene target site in subclone
3 讨 论
在植物遗传转化中,SMG起着非常重要的作用,可以帮助更准确快捷地得到转化子,但当转化完成后,SMG就失去了其使用价值。相反,在转基因植物中,SMG的存在被认为可能对人类健康和生态环境存在潜在的危害。随着转基因作物商业化进程的快速发展,无SMG转基因植物的研究和培育已成为国际上植物基因工程领域的一个趋势。目前,删除SMG的常用方法主要有:共转化系统法、DNA位点特异性重组系统法、转座子系统法、同源重组法和多元自主转化载体系统法[11]。利用这些方法在水稻[12-13]、玉米[14]、大豆[15]、柑橘[16]等作物研究上都成功实现了删除SMG。经过这么多年的发展,删除SMG的技术也在不断发展完善中,但目前这些技术大多处于实验室研究领域,且每种方法都存在着一定的局限性。因此简单快捷高效地删除SMG技术的开发和应用,仍然是国内外转基因研究领域的一项重要课题。
CRISPR/Cas9技术自2013年初建立以来,被认为是一种相比ZFN和TALEN操作更简单快捷、作用更高效准确的基因组编辑技术。目前已成功广泛应用于玉米[17]、水稻[18-20]、小麦[21-22]、大豆[23-24]、番茄[25]等作物转基因研究中,编辑效率有的甚至可以达到100 %。本研究利用CRISPR/Cas9技术,针对水稻转基因中常用的SMGhptII基因进行敲除。首先将构建的用于编辑hptII基因的编辑载体转入野生型水稻日本晴中,筛选获得Cas9表达的纯合株系;再将得到的编辑hptII基因的水稻纯合植株与含有hptII基因的转基因水稻植株进行杂交,在杂交F1代植株中即获得了hptII基因被敲除的植株。进一步,F1代植株通过自交得到F2代,在F2代中筛选可得到hptII基因DNA缺失、不能翻译出HPTII抗性蛋白,且不含有用于进行编辑hptII基因所转入水稻植株的外源DNA的植株,可以得到彻底删除转基因水稻中的hptII基因和其他非目的基因的外源片段的植株。
4 结 论
利用CRISPR/Cas9技术,成功获得了可用于敲除水稻转基因中hptII基因的转基因水稻,利用该水稻与含hptII基因的转基因水稻杂交,即可得到敲除hptII基因的水稻。利用该方法可在转化完成后才删除SMG,具有简单、通用的特点。
猜你喜欢
学与玩(2022年10期)2022-11-23
——一道江苏高考题的奥秘解读和拓展
中学生物学(2022年7期)2022-09-07
成都医学院学报(2022年4期)2022-08-19
今日农业(2022年3期)2022-06-05
中国农学通报(2022年12期)2022-06-01
中国糖料(2022年2期)2022-04-06
中国种业(2021年11期)2021-11-25
江西农业学报(2021年4期)2021-04-20
三农资讯半月报(2020年11期)2020-06-21
中学生物学(2019年7期)2019-10-17
