
法匹拉韦的合成工艺优化
2020-05-28 10:47邓玉晓赵思太张宗磊刘文涛段崇刚孙晋瑞
食品与药品 2020年2期
邓玉晓,张 宁,赵思太,张宗磊,刘文涛,段崇刚,孙晋瑞
(山东省药学科学院 山东省化学药物重点实验室,山东 济南 250101)
法匹拉韦(favipiravir,1),化学名称为6-氟-3-羟基吡嗪-2-甲酰胺,是日本富山化学工业公司研发的流感治疗药物[1],2011年3月在日本完成Ⅲ期临床试验[2],2014年3月被日本政府批准上市,商品名是Avigan[3]。法匹拉韦是一种病毒RNA聚合酶抑制剂,能阻断病毒RNA合成,从而起到抗病毒的作用;同时,法匹拉韦对哺乳动物细胞内的RNA合成不产生任何抑制作用,因此是一种安全有效的抗病毒药物。由于其特定的作用机制,除流感病毒外,法匹拉韦还可对抗其他多种RNA病毒,如HIV、黄热病、埃博拉等,有望应用于多种病毒感染的治疗,具有良好的市场前景[4]。
1的合成方法已有文献综述[5],有报道以2-氨基丙二酸二乙酯盐酸盐(2)为起始原料,与碳酸氢钠反应得2-氨基丙二酸二乙酯(3),与氨气的乙醇溶液反应3 d,得2-氨基丙二酰胺(4),与40 %乙二醛反应生成3-羟基吡嗪-2-甲酰胺(5),吡啶催化下,与溴素反应生成6-溴-3-羟基吡嗪-2-甲酰胺(6),在三乙胺的催化下,与三氯氧磷反应生成3, 6-二氯吡嗪-2-甲腈(7),在氟化钾作用下生成3, 6-二氟吡嗪-2-甲腈(8),在乙酸和三乙胺作用下发生3位水解,成盐生成6-氟-3-羟基吡嗪-2-甲腈二环己胺盐(9),最后在双氧水和氢氧化钠作用下生成1,总收率约9 %(以2计)[6]。但化合物3的2位氨基易与酯基发生氨酯交换,易导致化合物3变质,存储不便;制备4时,反应时间过长;制备6时,采用吡啶为催化剂,溴素为溴代试剂,吡啶具有恶臭且毒性大,溴素具有刺激性气味和强腐蚀性,反应后处理困难;制备7时,采用硅胶柱层析的方式纯化,操作过程繁琐,制备9时,采用成二环己胺盐的方式进行纯化,二环己胺具有渗透性臭气且毒性大,同时成盐这种提纯方式不符合绿色化学的理念。本文在参考上述文献合成路线的基础上对工艺作了进一步优化,提高了产品质量和工艺可操作性。具体合成路线见图1。

图1 化合物1的合成路线
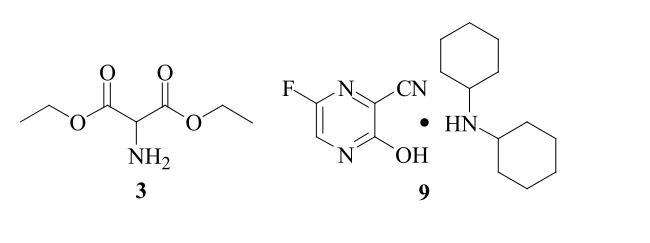
图2 化合物3和化合物9的结构
1 仪器与材料
1.1 仪器
Varian INOVA-300 核磁共振波谱仪(美国瓦里安公司);4000 Q TRAP 质谱仪(美国AB SCIEX公司);IKA HB 10 旋转蒸发仪(德国IKA);循环水式多用真空泵(郑州长城);DZF型真空干燥箱(北京市永光明医疗仪器厂);Agilent 1100高效液相色谱仪(美国安捷伦)。
1.2 药品与试剂
2-氨基丙二酸二乙酯盐酸盐(南京康满林,纯度>98.5 %);N-溴代丁二酰亚胺(上海阿拉丁,纯度>98.5 %);无水氟化钾(上海阿拉丁,纯度>98.5 %);四丁基溴化铵(河南万象,纯度>98.5 %);其余试剂均为分析纯。
2 方法与结果
2.1 2-氨基丙二酰胺(化合物4)的合成
将化合物2(1.0 kg,4.73 mol)、饱和氨气甲醇溶液(15.0 L)加至高压釜中,保持25~35℃,搅拌1 h,升温至65~70 ℃,搅拌3 h,降温至50 ℃,抽滤,滤液蒸除溶剂,得浅黄色固体4(506.0 g,收率91.5 %),mp 194.1~195.2 ℃(文献[6]:收率69 %,mp 198~200 ℃)。MS(m/z):118.0[M+H]+。
2.2 3-羟基吡嗪-2-甲酰胺(化合物5)的合成
将去离子水(3.0 L)加入反应瓶中,搅拌下加入氢氧化钠(70.2 g,1.8 mol),降温至40 ℃以内,依次加入85 %磷酸(99.9 g,0.9 mol)和4(506.0 g,4.3 mol),于20~30 ℃同时滴加8 mol/L氢氧化钠溶液(566.0 ml,4.5 mol)和40 %乙二醛水溶液(502.0 ml,4.5 mol),约1 h滴毕,保持20~30 ℃搅拌2 h,升温至85 ℃,滴入浓盐酸(474.2 ml,5.7 mol),逐渐降温冷却至10 ℃,搅拌反应1 h。抽滤,滤饼用去离子水(1.0 L)洗涤,60 ℃真空干燥,得褐色固体5(492.0 g,收率82.0 %),mp 261.2~263.3 ℃(文献[6]:收率64 %,mp 260~263 ℃)。MS(m/z):140.0[M+H]+;1H NMR(400 MHz,DMSO-d6)δ:12.69(s,1H,NH),11.15(s,1H,NH),7.93(s,1H,OH),7.87(d,J=3.2 Hz,1H,H-5),7.49(d,J=3.2 Hz,1H,H-6)。
2.3 6-溴-3-羟基吡嗪-2-甲酰胺(化合物6)的合成
将5(492.0 g,3.5 mol),DMF(1.0 L)加入反应瓶,加热至90 ℃,滴入NBS(640.8 g,3.58 mol)的DMF(1.0 L)溶液,控制滴加速度保持内温90~110 ℃。滴毕保温反应2 h,冷却至50~60℃,加入甲苯(1.0 L)和去离子水(3.0 L),冷却至15 ℃,抽滤,滤饼用水洗涤,60 ℃真空干燥,得土黄色固体6(587.0 g,收率76.0 %),mp 189.2~190.3 ℃(文献[6]:收率65 %,mp 189~191℃)。MS(m/z):216.0[M-H]-,218.0[(M+2)-H]-(1:1);1H NMR(400 MHz,DMSO-d6)δ:11.65(s,1H,OH),7.87(s,1H,H-5),6.31(s,2H,-CONH2)。
2.4 3, 6-二氯吡嗪-2-甲腈(化合物7)的合成
搅拌下向反应瓶中依次加入三氯氧磷(1652.0 g,10.8 mol)和6(587.0 g,3.4 mol),保持温度不高于80 ℃,滴加DIPEA(1044.0 g,8.1 mol),加毕升温至80~90 ℃反应2 h,升温至回流继续反应4 h,常压蒸馏直至没有馏分流出,将反应液降温至40 ℃,缓慢滴加 10 %碳酸钠水溶液,保持温度不高于60 ℃,调节体系pH 3~4,采用甲苯(500.0 ml×2)萃取,合并有机相,依次用水(500.0 ml)和饱和氯化钠溶液(500.0 ml)洗涤,回流分水3 h,直接投入下一步反应。
2.5 3, 6-二氟吡嗪-2-甲腈(化合物8)的合成
搅拌下向反应瓶中依次加入DMF(2.4 L),甲苯(2.6 L),无水氟化钾(392.0 g,6.7 mol),四丁基溴化铵(87.0 g,0.3 mol),加热至回流,保持回流分水2 h,降温至45 ℃,保持温度不高于55 ℃,滴加上一步制得的化合物7的甲苯溶液,滴毕,保持温度55~60 ℃反应3 h,加入去离子水(2.0 L),反应液采用甲苯(1.0 L×2)萃取,合并有机相,依次用水(500.0 ml)和饱和氯化钠水溶液(500.0 ml)洗涤,蒸除溶剂,得浅黄色固体8(236.0 g,62.0 %)。mp 58.1~58.5 ℃(文献:收率[7]63.6 %,mp[8]57.5~57.6 ℃)。MS(m/z):140[M-H]-;1H NMR(400 MHz,DMSO-d6)δ:8.85(s,1H,H-5)。
2.6 6-氟-3-羟基吡嗪-2-甲腈(化合物10)的合成
搅拌下向四口瓶中依次加入DMF(1.0 L),醋酸钾(343.5 g,3.5 mol)和3, 6-二氟吡嗪-2-甲腈(236.0 g,1.7 mol),保持温度25~35 ℃之间反应3 h,向反应体系中加入去离子水(1.5 L),用乙酸乙酯(500.0 ml×2)洗涤,滴加4.0 mol/L的盐酸,调节pH 2~3,用乙酸乙酯(500.0 ml×3)萃取,合并有机相,依次用去离子水(500.0 ml)和饱和氯化钠水溶液(500.0 ml)洗涤,蒸干溶剂,得淡黄色油状物,直接用于下一步反应。
2.7 法匹拉韦(化合物1)的合成
搅拌下向四口瓶中依次加入去离子水(1.0 L)和氢氧化钠(53.0 g,1.3 mol),降温至30 ℃以下,加入如上所得的10(184.0 g,1.3 mol),15~25℃滴加30 %双氧水(45.0 g,1.3 mol),滴毕继续反应2 h,升温至40~50 ℃,滴加浓盐酸(117.0 ml,1.4 mol),冷却至5 ℃,抽滤,滤饼用去离子水(300.0 ml)洗涤,再用丙酮(2.0 L)重结晶,得白色结晶性固体1(135.0 g,收率65 %)。mp 189.0~192.0 ℃(文献:收率[9]89.0 %,mp 187.0~191.0 ℃)。纯度99.9 %[HPLC面积归一化法:色谱柱:Intersil ODS-3,流动相:甲醇-乙腈-1 %冰醋酸(30:20:50),流速:1 ml/min,柱温:30 ℃,检测波长:238 nm]MS(m/z):156.0[M-H]-;1H NMR(400 MHz,DMSO-d6)δ:12..65(s,1H,OH),8.25(s,1H,H-5),7.58~7.62(s,2H,-CONH2);13C NMR δ:126.5(C-5),139.5(C-2),141.0(C-6),157.5(C-3),169.7(-CONH2)。
3 讨论
3.1 制备4时,用甲醇代替乙醇为反应溶剂,梯度升温,先在25~35 ℃下搅拌1 h,使3游离出来,然后升温至65~70 ℃,在近乎均相的条件下反应,有效碰撞增多,反应时间由3 d缩短为3 h,收率由文献的69 %升高至91.5 %。
3.2 制备6时,本研究改用NBS和DMF,操作简单,反应收率由49 %提高至86 %。
3.3 化合物7具有强烈的致敏性,皮肤接触后会导致疱疹,痒痛难忍,一般持续3周以上才能痊愈,同时化合物7的熔点较低,容易升华,干燥过程中劳动保护困难。本研究采用甲苯共沸除水法干燥该化合物,体系中水分低于0.1 %时直接投入反应,有效避免合成人员的接触危害。
3.4 制备9时,采用成盐洗涤法,在调酸析晶前,采用甲苯洗涤反应液,能有效去除有机杂质,缩减成本,简化操作。
4 结论
以2-氨基丙二酸二乙酯盐酸盐为起始物料经过氨解、成环、溴代、氯代、脱水、氟代、取代、水解等步骤合成法匹拉韦,通过MS、1H NMR等方法进行结构确证,产品纯度达99.5 %以上,总收率23.8 %,合成工艺操作简单,收率高,具有工业化应用前景。
猜你喜欢
中国防痨杂志(2022年1期)2022-11-24
江苏农业科学(2022年19期)2022-10-28
盐科学与化工(2022年6期)2022-06-20
氯碱工业(2021年6期)2021-12-25
今日健康(2021年11期)2021-12-09
医疗装备(2021年11期)2021-12-04
中国防痨杂志(2021年12期)2021-12-04
酿酒科技(2021年11期)2021-11-24
能源工程(2021年2期)2021-07-21
分析化学(2019年3期)2019-03-30
