
薏苡仁中药饮片指纹图谱的构建和识别模式研究
2020-05-28 10:47毕天琛杨国宁仝桂平赵丹彤刘延娟
食品与药品 2020年2期
毕天琛,杨国宁,仝桂平,赵丹彤,刘延娟
(1.菏泽市食品药品检验检测研究院,山东 菏泽 274000;2.山东中医药大学 药学院,山东 济南 250355)
薏苡仁为禾本科植物薏苡[Coix lacryma-jobiL.var.mayuen(Roman.) Stapf]的干燥成熟种仁[1],收载于2015年版《中国药典》(一部),是我国传统的药食两用中药,具有较高的经济价值、营养价值和药用价值[2],被誉为“世界禾本科植物之王”[3]。
总量统计矩法是将指纹图谱作为众多高斯曲线叠加而成的概率密度曲线,用统计学方法来分析指纹图谱的内在特征,按一维随机向量统计原理,用总量零阶矩(AUCT)、总量响应率(AUCPWT)、总量一阶矩(MCRTT)、总量二阶矩(VCRTT)4个参数进行全面的定性定量研究[4]。
目前,已有采用高效液相色谱(HPLC)和气相色谱质谱联用(GC-MS)等方法建立薏苡仁指纹图谱的相关研究报道[5-8],但均未进行系统的信息综合处理分析。本文采用HPLC建立薏苡仁化学指纹图谱后进行总量统计矩分析,并采用夹角余弦法和相关系数法进行相似度评价,结合聚类分析和主成分分析对18批次薏苡仁饮片进行指纹图谱识别和分类,为薏苡仁质量评价及标准的制定提供科学依据。
1 仪器与试药
1.1 仪器
安捷伦1200高效液相色谱仪(美国安捷伦公司);PL-ELS 2100蒸发光散射检测器(Polymer Laboratories公司);XS105电子天平(梅特勒-托利多);AB104-S电子天平(梅特勒-托利多);FRQ-1010T超声波清洗器(杭州法兰特); Milli-Q Advantage A10超纯水仪(默克密理博公司)
1.2 试药
甘油三油酸酯对照品(中国食品药品检定研究院,批号:111692-201806,含量99.9 %);乙腈(色谱纯,德国默克公司);二氯甲烷(色谱纯,德国默克公司)。本实验收集18批次市售不同产地的薏苡仁饮片,并经菏泽市食品药品检验检测研究院马海春研究员鉴定,详见表1。
2 方法与结果
2.1 色谱条件
色谱柱:安捷伦C18柱(250 mm×4.6 mm,5 μm);流动相:乙腈-二氯甲烷(65:35);柱温30 ℃;流速1 ml/min;蒸发光散射检测器参数:漂移管温度50 ℃;载气体积流量为0.6 L/min。

表1 18批薏苡仁饮片来源
2.2 溶液的配制
2.2.1 供试品溶液的制备 称取薏苡仁粉末(过三号筛)约0.6 g,精密称定,置具塞锥形瓶中,精密加入流动相50 ml,称定重量,浸泡2 h,超声处理(功率300 W,频率50 kHz)30 min,放冷,再称定重量,用流动相补足减失的重量,摇匀,滤过,取续滤液作为供试品溶液。
2.2.2 对照品溶液的制备 精密称取甘油三油酸酯对照品适量,置于量瓶中,加流动相溶解并稀释,制备成甘油三油酸酯浓度为0.14 mg/ml的对照品溶液。
2.3 方法学考察
2.3.1 精密度试验 取同一薏苡仁供试品溶液,按2.1项下色谱条件,连续进样测定6次,以甘油三油酸酯(6号峰)为参照峰,计算各共有峰的相对保留时间与相对峰面积。结果各相对保留时间的RSD均小于0.56 %,各相对峰面积的RSD均小于2.05 %,表明仪器精密度良好。
2.3.2 稳定性试验 取同一薏苡仁供试品溶液,按2.1项下色谱条件分别于0,2,4,6,8,12 h进样测定,以甘油三油酸酯(6号峰)为参照峰,计算测得各共有峰的相对保留时间与相对峰面积。结果各相对保留时间的RSD均小于0.61 %,各相对峰面积的RSD均小于2.42 %,表明供试品溶液在12 h内稳定。
2.3.3 重复性试验 取同一薏苡仁供试品6份,精密称定,分别按2.2.1项下方法制备供试品溶液,按2.1项下色谱条件测定,以甘油三油酸酯(6号峰)为参照峰,计算测得各共有峰的相对保留时间与相对峰面积。结果各相对保留时间的RSD均小于0.88 %,各相对峰面积的RSD均小于2.61 %,表明方法的重复性良好。
2.4 指纹图谱的建立
2.4.1 指纹图谱共有模式的建立 将18个不同批次薏苡仁样品HPLC色谱图分别导入“中药色谱指纹图谱相似度评价系统(2012A)”软件,选定S1号样品色谱图为参照图谱,采用中位数法自动匹配生成对照图谱,结果见图1。由图1可知,共获得7个共有峰,参照文献报道进行定位[6-9],指认1~7号色谱峰依次为三亚油酸甘油酯、1, 2-亚油酸-3-油酸甘油酯、3-棕榈酸二亚油酸甘油酯、1, 2-油酸-3-亚油酸甘油酯、棕榈酸亚油酸油酸甘油酯、甘油三油酸酯、棕榈酸二油酸甘油酯。其中6号峰(甘油三油酸酯)峰面积所占比例较大且相对稳定,分离度较好,因此确定其为参照峰(S)。
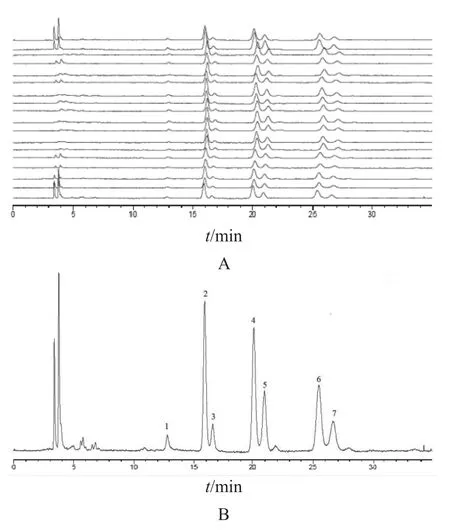
图1 18批薏苡仁样品的叠加HPLC指纹图谱(A)与对照指纹图谱(B)
2.4.2 共有峰相对保留时间与相对峰面积 以各批次的6号峰为参照峰(甘油三油酸酯),计算同一样品图谱中各共有峰的相对峰面积,结果见表2。由表2可见,相对峰面积波动较大,RSD范围为0~27.126 %,表明薏苡仁指纹图谱中主要峰群的整体面貌基本一致,但各成分含量差别较大。

表2 各共有峰的相对峰面积
2.4.3 总量统计矩分析 采用总量统计矩分析法计算总量零阶矩(AUCT)、总量响应率(AUCPWT)、总量一阶矩(MCRTT)、总量二阶矩(VCRTT)[10]等总量统计矩参数,根据计算结果评价不同批次薏苡仁样品的差异,结果见表3。

表3 18批次薏苡仁样品的总量统计矩参数
由表3可见,不同产地薏苡仁样品的指纹图谱的总量统计矩参数差异明显,表明不同产地薏苡仁在“量”上具有一定的差异性,就总量响应率(AUCPWT)而言,S17最大,S4最小。
2.4.4 相似度评价 分别采用夹角余弦法和相关系数法[11]对18批薏苡仁样品的HPLC指纹图谱进行相似度分析,结果见表4。夹角余弦法计算的相似度均在0.97以上,相关系数法计算的相似度均在0.98以上。两种相似度评价结果均表明各批次薏苡仁样品的质量相对稳定,具有较高的相似度。
2.5 聚类分析
采用SPSS 22.0软件,以18批次薏苡仁饮片的7个共有峰峰面积数据为变量,选用组间联接法,Euclidean距离为测度,进行聚类分析,结果见图2。18批次样品可聚为3类,其中S17 为单独一类,S1、S6、S9、S12、S16、S18聚为一类,其余样品聚为一类。观察各批次样品共有峰相对峰面积可发现,S17中7个色谱峰面积均高于其余样品,含量均为最高,说明其整体质量相对较好。

表4 夹角余弦法和相关系数法计算不同产地薏苡仁样品的相似度

图2 18批次薏苡仁样品聚类分析树状图
2.6 主成分分析
将指纹图谱中7个共有峰的峰面积数据为变量,构建18×7的原始数据矩阵,采用SPSS22.0 软件,对18批次薏苡仁样本进行主成分分析。主成分个数提取原则为主成分的特征值大于1的前m个主成分,结果见表5、表6。

表5 薏苡仁主成分分析的特征值与方差贡献率结果
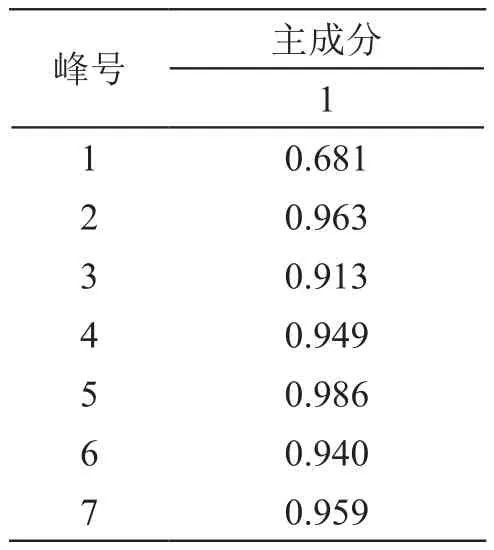
表6 薏苡仁主成分载荷矩阵
由表5可知,提取到了1个主成分,特征值为5.900,方差贡献率为84.283%。由表6可见,2~7号峰在第一种主成分上载荷均大于0.91,说明2~7号峰成分对薏苡仁的质量贡献较大,其含量的高低影响薏苡仁药材质量的优劣。
以上述1个主成分因子为变量绘制得分图,见图3。18批次样品可归为3组,S17样品为一组,S9、S12、S16、S1、S18和S6为一组,其余样品为一组。与聚类分析结果相近;但在S6的分组上,两种分析结果略有差异。

图3 不同批次薏苡仁样品主成分得分散点图
3 讨论
3.1 实验条件的优化
本研究在供试品溶液的制备过程中,分别考察了甲醇-二氯甲烷(60:40)、含10 %,20 %,35 %,50 %二氯甲烷的乙腈溶液5种提取溶剂及超声、回流两种提取方法的提取效果,结果以乙腈-二氯甲烷(65:35)为提取液,先静置2 h,再超声处理30 min钟提取的供试品溶液色谱图效果最好,且处理方法简便。考察了不同柱温、漂移管温度和载气体积流量下峰形的变化,发现柱温为30 ℃,漂移管温度为50 ℃,载气体积流量为0.6 L/min时,色谱峰保留时间、峰形和分离度较理想。
3.2 薏苡仁饮片指纹图谱识别
本研究采用HPLC-ELSD建立了18批次薏苡仁饮片的指纹图谱,共标定7个共有峰,并通过文献比对指认7个共有峰分别为三亚油酸甘油酯、1, 2-亚油酸-3-油酸甘油酯、3-棕榈酸二亚油酸甘油酯、1, 2-油酸-3-亚油酸甘油酯、棕榈酸亚油酸油酸甘油酯、甘油三油酸酯、棕榈酸二油酸甘油酯。
在指纹图谱识别模式中采用总量统计矩分析法计算出各批次样品的总量统计矩参数,结果显示,各批次样品的总量统计矩参数差异较大,其中S17 值偏大,S1、S6、S9、S12、S16、S18值较相近,其余样品值较相近,其结果与聚类分析和主成分分析结果一致。
4 结论
本研究采用相似度评价、聚类分析与主成分分析法对各批次样品进行分类识别分析,结果表明各批次样品在定性分析中具有较高的相似性,但不同批次样品在7种成分含量上有较大差异。主成分分析发现2~7号色谱峰对薏苡仁的质量贡献均大于0.9,说明这些成分的含量高低对薏苡仁质量影响较大,提示薏苡仁饮片质量控制局限于甘油三油酸酯单一检测,不能全面有效地反应样品整体质量状况,应进一步研究其余5种成分对其质量的影响。综上所述,本研究建立的薏苡仁饮片HPLC指纹图谱,结合总量统计矩分析、相似度分析、聚类分析和主成分分析法,可较系统全面地反映薏苡仁饮片的整体质量状况。
猜你喜欢
生物技术进展(2022年5期)2022-10-11
天津药学(2022年4期)2022-09-14
药学实践杂志(2022年3期)2022-05-27
中草药(2022年10期)2022-05-24
新农业(2020年18期)2021-01-07
现代农村科技(2020年2期)2020-03-24
环球时报(2018-03-01)2018-03-01
中学科技(2017年11期)2017-12-26
中学生数理化·八年级物理人教版(2015年10期)2016-01-04
科技视界(2015年4期)2015-01-02
