
原核表达载体pET28a-3×flag-LukS-PV的构建与表达鉴定
2020-03-02 08:57汪自然王杨燕马筱玲
山西医科大学学报 2020年1期
汪自然,马 凡,王杨燕,马筱玲
(安徽医科大学附属省立医院检验科,合肥 230001;*通讯作者,E-mail:xiaolingma@126.com)
金黄色葡萄球菌是临床上常见的引起感染的病原体,部分金黄色葡萄球菌可编码产生PV-杀白细胞素(Panton-Valetine leukocidin,PVL)[1,2]。美国CDC于1999年首次报道了PVL阳性的金黄色葡萄球菌导致了4例儿童患者脓毒血症死亡,引起临床广泛关注[3]。LukS-PV和LukF-PV两个组分共同组成PVL,LukS-PV可与白细胞细胞膜上的受体结合,随后LukF-PV结合引发白细胞裂解[4]。近年来,我们发现单组分LukS-PV可以诱导髓系白血病细胞凋亡和分化[5],有望成为一种新的肿瘤靶向杀伤药物,具有深入研究价值。但是LukS-PV的具体调控分子机制仍不清楚,本研究通过构建pET28a-3×flag-LukS-PV原核表达载体并进行表达纯化及鉴定,为后续研究LukS-PV与其他分子的相互作用及具体机制奠定了基础。
1 材料与方法
1.1 材料
1.1.1 质粒与菌株 pET28a质粒,大肠杆菌感受态DH5α、BL21,产PVL的金黄色葡萄球菌铜-23均由本实验室保存。p3×flag-myc-CMV-24质粒由中国科技大学附属第一医院妇产科实验室惠赠。
1.1.2 主要试剂 细菌基因组DNA提取试剂盒、质粒小提试剂盒、胶回收试剂盒购于北京天根公司;限制性核酸内切酶NdeⅠ、BamHⅠ、XhoⅠ,T载体pMD-18T,Taq酶购于日本Takara公司;SDS-PAGE胶、快速封闭液购于上海雅酶生物技术公司;抗His单克隆抗体购于美国CST公司;抗flag多克隆抗体、羊抗兔IgG购于武汉Abclonal公司;BCA试剂盒购于上海碧云天公司。
1.2 方法
1.2.1 引物设计与基因扩增 按照NCBI上的基因序列,使用Primer Premier 5.0设计引物。LukS-PV上游引物(F):5′-ACGCGGATCCGAATC TAAAG CTGATAACAATATTGAGAATATTG-3′(下划线为BamHⅠ酶切位点);LukS-PV下游引物(R):5′-ACCGCTCGAGTCAATTAT GTCCTTTCACTTTAA TTTCATGAG-3′(下划线为XhoⅠ酶切位点)。3×flag上游引物(F):5′-CGCCATATGGCGGACTACAAAGACCATGACG-3′(下划线为NdeⅠ酶切位点);3×flag下游引物(R):5′-CGCGGATCCGCGCTTGTCATCGTCATCCTT G-3′(下划线为BamHⅠ酶切位点),引物由安徽通用生物公司合成。分别以产PVL的金黄色葡萄球菌铜-23的DNA和p3×flag-myc-CMV-24质粒为模板,扩增LukS-PV和3×flag序列。具体PCR条件为:94 ℃预变性5 min,94 ℃变性30 s,56 ℃退火30 s,72 ℃延伸1 min,共30个循环,72 ℃延伸7 min,4 ℃冷却5 min。PCR体系为2×PCR mix 20 μl,模板2 μl,上下游引物各1 μl,灭菌ddH2O 16 μl。PCR产物经10 g/L琼脂糖凝胶电泳后使用胶回收纯化试剂盒切胶回收备用。
1.2.2 pET28a-3×flag质粒的构建与鉴定 将回收后的3×flag片段和LukS-PV片段分别与T载体pMD-18T于16 ℃过夜连接,具体连接体系为回收片段4 μl,pMD-18T 1 μl,SolutionI 5 μl。次日将连接产物转化至大肠杆菌DH5α中,均匀涂布于含有氨苄抗生素平板上。取平板上单菌落摇菌提质粒,并使用PCR初步鉴定后送至安徽通用生物公司测序。测序鉴定无误后的pMD-18T-3×flag质粒和pET28a质粒分别使用限制性核酸内切酶NdeⅠ和BamHⅠ进行双酶切,酶切条件为37 ℃ 2 h。双酶切后使用琼脂糖凝胶电泳分离,分别回收pMD-18T-3×flag质粒的小片段(目的片段)和pET28a的大片段(载体片段),二者按照4 ∶1的物质的量比混合加入5 μl的Solutin I于16 ℃过夜连接。次日转化至感受态大肠杆菌DH5α中,经卡那抗生素平板筛选后,取单菌落摇菌提质粒,PCR扩增目的基因鉴定后送安徽通用生物公司测序鉴定。
1.2.3 pET28a-3×flag-LukS-PV质粒的构建与鉴定 取经测序鉴定无误的pMD-18T-LukS-PV质粒和pET28a-3×flag质粒,使用限制性核酸内切酶BamHⅠ和XhoⅠ双酶切,电泳后分别回收pMD-18T-LukS-PV质粒的小片段和pET28a-3×flag质粒的大片段,按照4 ∶1的物质的量比混合后加入5 μl的Solution I于16 ℃过夜连接。次日转化至感受态大肠杆菌DH5α中,经卡那抗生素平板筛选后,取单菌落摇菌提质粒,PCR扩增目的基因鉴定后送安徽通用生物公司测序鉴定。将鉴定无误的pET28a-3×flag-LukS-PV质粒转化入感受态大肠杆菌BL21中保存备用。
1.2.4 重组蛋白的诱导和纯化 取构建好的含有pET28a-3×flag-LukS-PV质粒的BL21大肠杆菌单菌落过夜摇菌活化,次日按1 ∶100的比例加入200 ml的LB培养基中,并加入200 μl的卡那抗生素。37 ℃ 220 r/min至菌液的OD600达到0.6-0.8(对数生长期)后,取1 ml诱导前菌液作对照,加入诱导剂IPTG至终浓度为0.8 mmol/L,诱导条件为37 ℃ 220 r/min。分别于诱导后1,2,3,4,5,6 h取样,6 h后离心收集细菌,去除培养基,用20 ml PBS重悬,超声破碎后分别留取上清液和沉淀。超声破碎后上清液采用His纯化试剂盒纯化,纯化后得到的蛋白产物使用BCA法进行蛋白定量测定。分别取诱导前,诱导后1-6 h,诱导6 h细菌超声破碎后上清液、沉淀,纯化后蛋白产物使用10%的SDS-PAGE分析表达量。
1.2.5 重组蛋白的Western blot鉴定 取诱导前和纯化后蛋白产物使用10%的SDS-PAGE电泳分离后,电泳条件为先80 V至marker分离开后120 V溴酚蓝到达玻璃板底部,电泳结束后剥胶转至NC膜上,转膜条件为200 mA 90 min。用雅酶公司快速封闭液封闭1 h,分别以兔抗His标签抗体(1 ∶1 000稀释)和鼠抗flag标签抗体(1 ∶5 000稀释)为一抗,于4 ℃摇床过夜孵育。次日TBST洗去未结合的一抗,分别以羊抗兔(1 ∶5 000稀释)IgG和羊抗鼠(1 ∶5 000稀释)IgG作为二抗,孵育2 h后TBST洗去未结合的二抗,加入ECL显影剂于曝光成像仪下显像。
2 结果
2.1 LukS-PV和3×flag基因的PCR扩增结果
PCR扩增LukS-PV和3×flag基因后,经10 g/L琼脂糖凝胶电泳分离后显示,LukS-PV基因约在867 bp处有明显亮带,3×flag基因约在84 bp处有明显亮带(见图1),与预测相符。
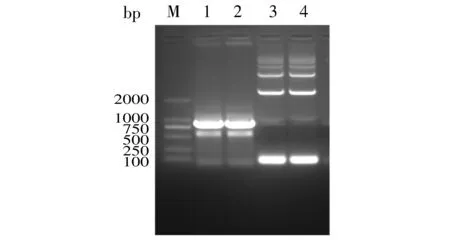
M. DL2000 DNA marker; 1,2. LukS-PV基因扩增产物; 3,4. 3×flag基因扩增产物图1 PCR扩增目的基因结果图Figure 1 Results of PCR amplification of target genes
2.2 pET28a-3×flag-LukS-PV载体的鉴定
对pMD-18T-LukS-PV质粒使用限制性核酸内切酶BamHⅠ、XhoⅠ双酶切鉴定,对pMD-18T-3×flag质粒使用限制性核酸内切酶NdeⅠ、BamHⅠ双酶切鉴定,结果见图2。以pET28a-3×flag-LukS-PV为模板,进行PCR扩增鉴定,可扩增出LukS-PV和3×flag特异性条带(见图3)。pET28a-3×flag-LukS-PV质粒以通用引物(T7,T7ter)双向测序,将目的基因序列与NCBI数据库中比对,显示100%配对,无碱基突变。部分测序图见图4,质粒图谱见图5。
2.3 重组蛋白的诱导表达
分别取诱导前、加入IPTG诱导后1-6 h、超声破碎后沉淀、上清、纯化后蛋白作SDS-PAGE电泳分析,结果见图6。由图6可见,诱导后在约40 kD处出现蛋白条带,而诱导前此处无条带。超声破碎后上清中蛋白表达量较沉淀中更高,表明其为可溶性表达。使用BCA试剂盒测定纯化后重组蛋白浓度,蛋白浓度为2.37 mg/ml。SDS-PAGE电泳显示,纯化后蛋白条带浓集,杂带较少,浓度和纯度均较高。
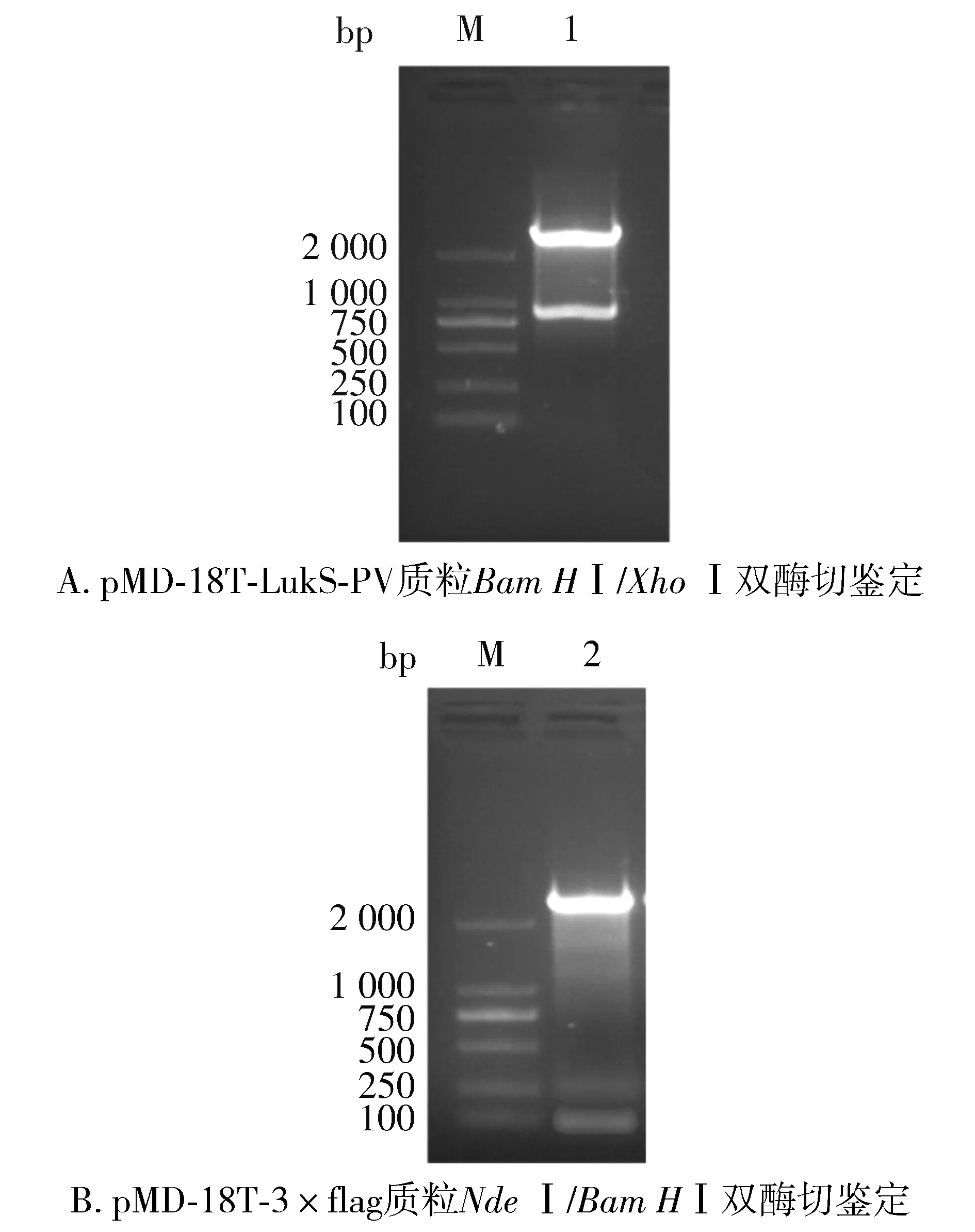
M. DL2000 DNA marker; 1. pMD-18T-LukS-PV质粒Bam HⅠ/Xho Ⅰ双酶切; 2. pMD-18T-3×flag质粒Nde Ⅰ/Bam HⅠ双酶切图2 重组T载体双酶切鉴定图Figure 2 Double enzyme digestion and identification of recombinant T vector
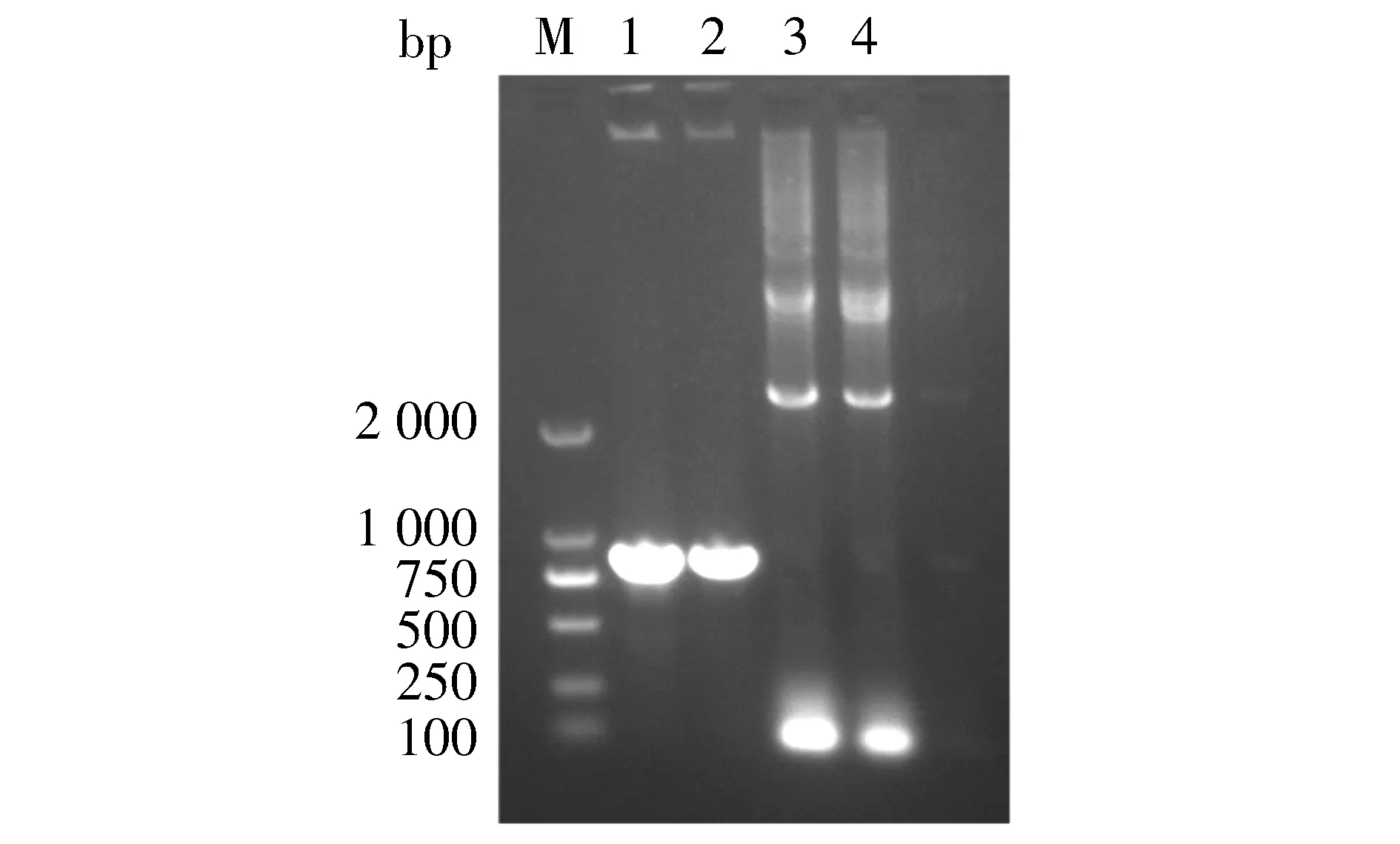
M. DL 2000 DNA marker; 1,2. pET28a-3×flag-LukS-PV扩增LukS-PV基因; 3,4. pET28a-3×flag-LukS-PV扩增3×flag基因图3 pET28a-3×flag-LukS-PV质粒的PCR鉴定Figure 3 PCR identification of pet28a-3×flag-LukS-PV plasmid
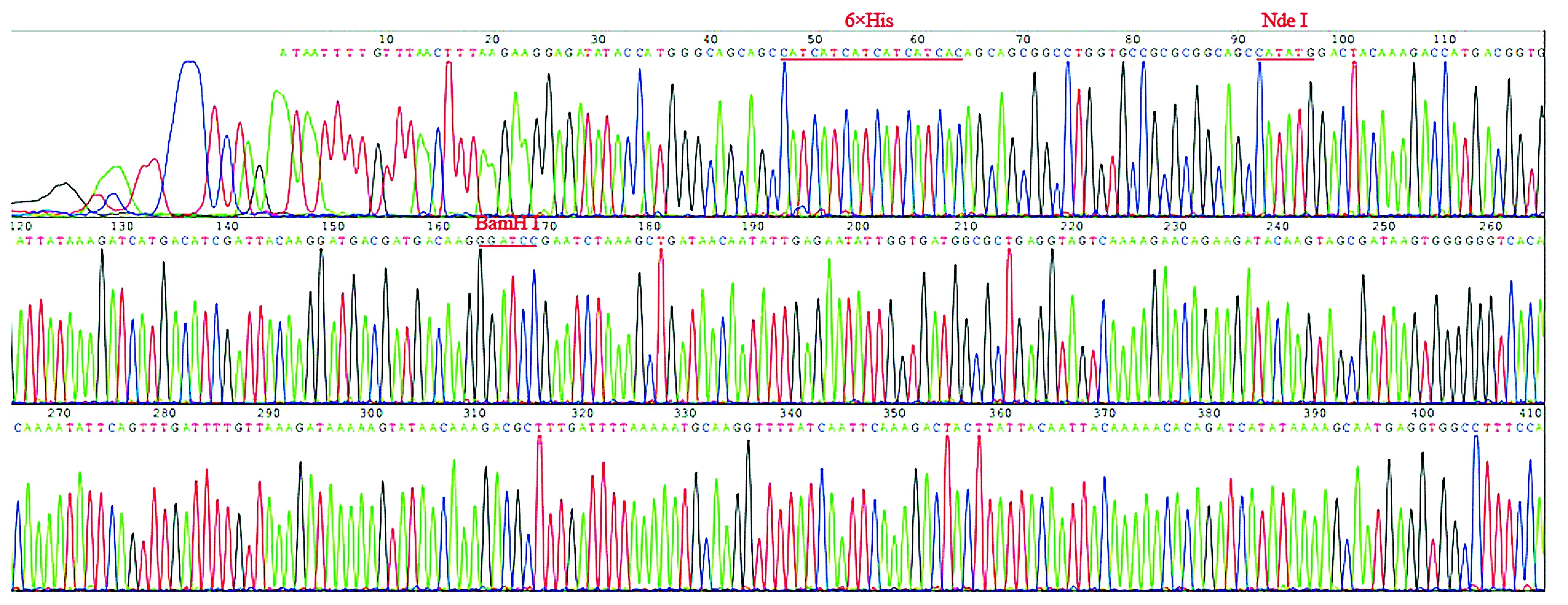
图4 pET28a-3×flag-LukS-PV质粒的部分测序图Figure 4 Partial sequencing diagram of pet28a-3×flag-LukS-PV plasmid
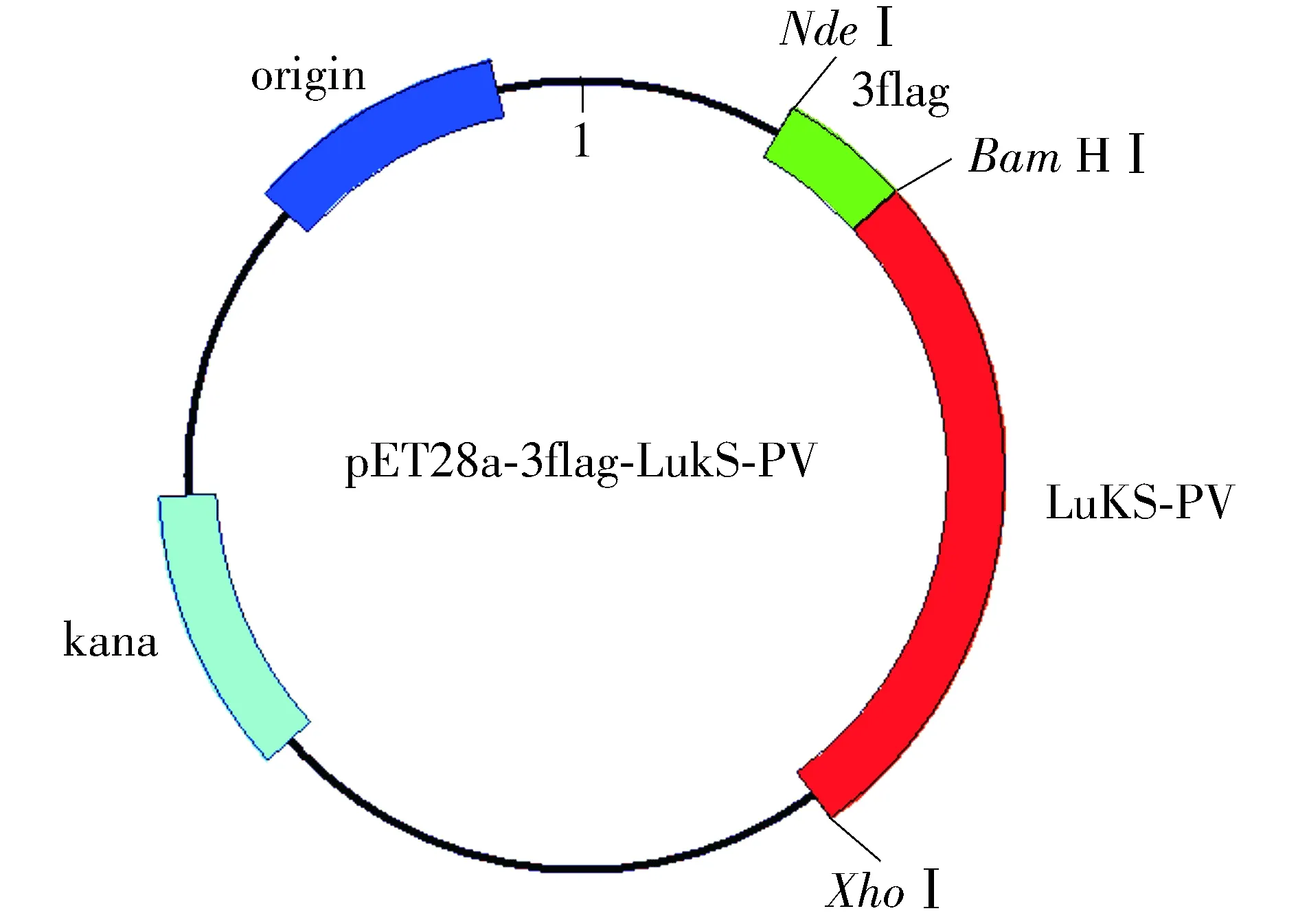
图5 pET28a-3×flag-LukS-PV质粒图谱Figure 5 Plasmid profile of pet28a-3×flag-LukS-PV
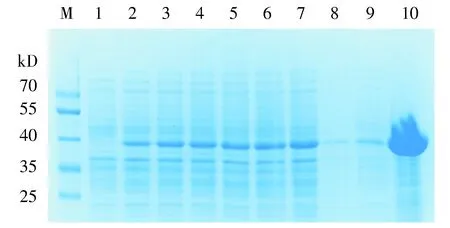
M.蛋白质marker; 1.pET28a-3×flag-LukS-PV诱导前; 2-7.pET28a-3×flag-LukS-PV经IPTG诱导1-6 h; 8.诱导6 h细菌超声破碎后沉淀; 9.诱导6 h细菌超声破碎后上清液; 10.纯化后蛋白产物图6 重组蛋白的表达量分析Figure 6 Expression analysis of recombinant protein
2.4 重组蛋白的Western blot鉴定
分别使用抗His单克隆抗体和抗flag多克隆抗体为一抗,对纯化后重组蛋白进行Western blot鉴定,结果如图7,与诱导前对比发现,纯化后蛋白在约40 kD处有特异性浓集条带。
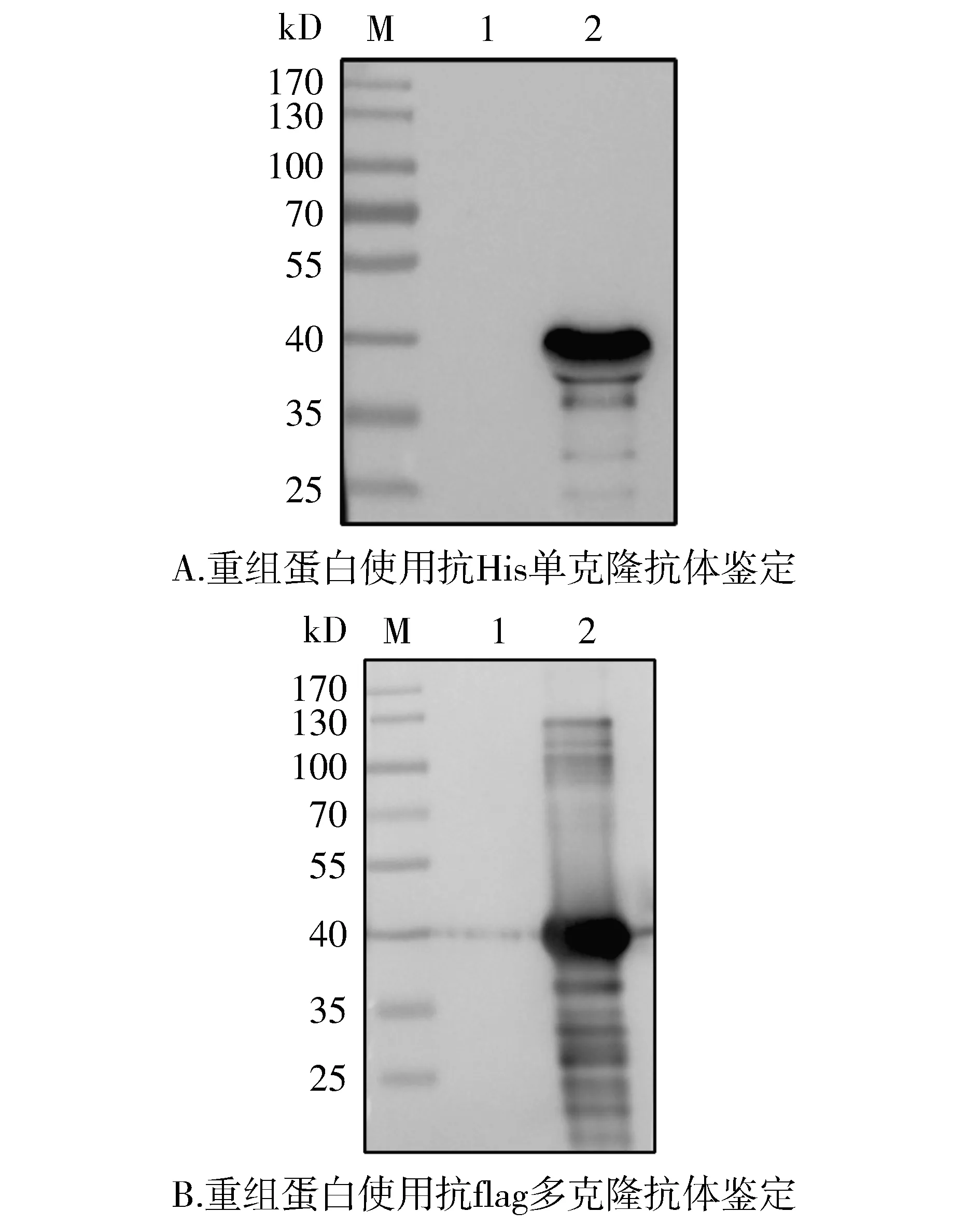
M. 蛋白质marker; 1. pET28a-3×flag-LukS-PV诱导前; 2. 纯化后蛋白产物图7 重组蛋白的Western blot鉴定Figure 7 Identification of recombinant protein by Western blot
3 讨论
LukS-PV是金黄色葡萄球菌杀白细胞素PVL的S组分,前期研究[5,6]表明LukS-PV诱导人白血病细胞系凋亡和周期阻滞,并通过动物实验进行验证,表明了LukS-PV有望成为一种新的肿瘤靶向治疗药物。3×flag是一段多肽蛋白标签,由22个氨基酸组成,其氨基酸序列为DYKDHDGDYKDHDIDYKDDDDK。3×flag标签在Western blot中检测较为敏感,且不会对融合蛋白的定位和功能产生影响[7]。3×flag作为分子标签与目的蛋白组成融合蛋白,用于观察蛋白定位及与其他分子之间的相互作用,具有广阔的应用价值。
常见的蛋白表达系统有原核(大肠杆菌)系统、哺乳动物表达系统、酵母表达系统、昆虫表达系统等[8]。大肠杆菌表达系统作为经典的蛋白表达系统,已成为外源蛋白表达的首选。蛋白表达纯化过程中,需要对温度、转速、IPTG浓度、诱导菌OD值等条件进行摸索,以获得重组蛋白的可溶性表达,避免变复性过程对蛋白的活性和功能影响。高玉录等[9]将LukS-PV基因克隆到Rosetta(DE3)plys宿主菌中,重组LukS-PV蛋白主要存在于包涵体中。常文娇等[10]通过去除LukS-PV基因的引导肽,降低了引导肽对蛋白表达的影响,促进了重组的可溶性表达。王青元等[11]构建了pET22b-3×flag-HPV18E6融合表达载体,并在大肠杆菌中诱导表达出融合重组蛋白。
本研究在课题组前期研究基础上通过PCR技术扩增得到LukS-PV和3×flag基因,并首先克隆到T载体pMD-18T载体上(TA克隆),以便于后续双酶切。通过改造载体,利用pET28a上不同位置的酶切位点,分步将LukS-PV和3×flag基因克隆至pET28a上,构建pET28a-3×flag-LukS-PV载体(图5),并转至大肠杆菌BL21宿主菌中。IPTG诱导后,可以纯化得到浓度和纯度均较高的重组蛋白,经Western blot验证与抗His抗体和抗flag抗体均有良好的反应性,可以用于后续的免疫共沉淀等实验。本研究成功纯化得到了高浓度和高纯度的3×flag-LukS-PV重组蛋白,为后续研究LukS-PV的生物学功能和具体机制奠定了基础。
猜你喜欢
——一道江苏高考题的奥秘解读和拓展
中学生物学(2022年7期)2022-09-07
成都医学院学报(2022年4期)2022-08-19
中国农学通报(2022年12期)2022-06-01
中国糖料(2022年2期)2022-04-06
湖南畜牧兽医(2021年6期)2022-01-24
中国种业(2021年11期)2021-11-25
食品安全导刊(2021年21期)2021-08-30
猪业科学(2021年5期)2021-06-02
检验医学(2021年4期)2021-04-28
浙江农业学报(2021年1期)2021-01-28
