
TRPM2在H9N2猪流感病毒感染小鼠PMVEC损伤中的表达与作用*
2019-12-25 06:10高晶萍张雅琪李珮瑶王少华张瑞华
中国病理生理杂志 2019年12期
罗 强, 高晶萍, 梁 亭, 张雅琪, 李珮瑶, 王少华, 李 军, 张瑞华, 徐 彤, △
(河北北方学院 1预防兽医学重点实验室, 2生命科学研究中心, 3基础医学院, 河北 张家口 075000)
瞬时受体电位阳离子通道M2(transient receptor potential cation channel M2,TRPM2)作为氧化应激敏感性离子通道在某些疾病致病机制中发挥重要作用。TRPM2参与肿瘤、炎症、缺血再灌注损伤以及糖尿病等致病过程[1]。氧化应激作用通过TRPM2-PLCγ1-PKCα信号通路弱化了呼吸道上皮细胞紧密连接蛋白的连接功能[2]。此外,肺泡上皮细胞TRPM2参与博来霉素诱导肺损伤炎症过程[3]。因此,越来越多的研究表明TRPM2作为细胞内部氧化还原反应的传感器启动氧化应激/活性氧类,进而导致内皮细胞通透性增强和凋亡过程,TRPM2可以作为氧化应激性疾病潜在的治疗靶点[4-5]。
近年来,动物源性流感病毒不断突破种属屏障并导致人类感染,已成为威胁人类健康的重要病原之一。目前研究显示流感病毒感染引起肺损伤的主要机制与氧化应激损伤有关。已有研究指出,氧化应激损伤和TLR4在H5N1禽流感病毒诱导急性肺损伤中发挥重要作用[6]。在研究H9N2猪流感病毒(swine influenza virus, SIV)诱导肺损伤机制时,本课题组发现氧化应激是导致急性肺损伤发生的重要原因之一[7-8]。鉴于氧化应激在流感病毒致病机制中的重要作用以及TRPM2可能是氧化应激性疾病的潜在治疗靶点,本研究探讨H9N2-SIV感染小鼠肺微血管内皮细胞(pulmonary microvascular endothelial cells, PMVEC)过程中TRPM2的表达与作用,为通过TRPM2有效干预/治疗流感病毒诱导的肺损伤提供参考资料。
材 料 和 方 法
1 材料
A/swine/HeBei/012/2008/(H9N2)病毒由河北北方学院预防兽医学重点实验室分离鉴定,其部分基因序列参见GenBank,序列号为CY063662、CY063663、CY063664和CY063665。该病毒经10日龄SPF鸡胚传代,收集含毒尿囊液,经红细胞凝集实验测定病毒血凝价,将效价在8 Log2以上含毒尿囊液进行分装、于-70 ℃冰箱保存,待用。小鼠PMVEC购自武汉普诺赛生命科技有限公司。
2 主要试剂
DMEM-H培养液、丙酮酸钠、L-谷氨酰胺和青-链霉素购自Gibco;胎牛血清(fetal bovine serum, FBS) 购自Corning;超氧化物歧化酶(superoxide dismutase, SOD)活性检测试剂盒、谷胱甘肽过氧化物酶(glutathione peroxidase, GSH-Px)检测试剂盒、活性氧簇(reactive oxygen species, ROS)检测试剂盒、总一氧化氮合成酶(nitric oxide synthase, NOS)定量试剂盒、ECL试剂盒、抗GAPDH抗体、BCA蛋白定量试剂盒和总ATP酶定量试剂盒购自凯基生物公司;羊抗兔IgG购自Invitrogen;抗TRPM2抗体购自Abcam。Corning Transwell培养板购自上海坤肯生物化工有限公司。
3 主要方法
3.1PMVEC培养 将PMVEC使用含10% FBS、L-谷氨酰胺、丙酮酸钠和青-链霉素的DMEM-H培养液,培养至对数生长期使用。
3.2H9N2-SIV感染 参照李珮瑶等[9]报道方法进行。取PMVEC已长成单层的24孔细胞培养板,弃去细胞培养液,PBS洗3次(每次3 min),1~3列加入10-2稀释H9N2-SIV液500 μL,4~6列加入相同体积的DMEM培养液,置37 ℃、5% CO2温箱中吸附2 h(中间振荡20 s)后,弃去病毒悬液或培养液,1~6列分别加入含0.3 mg/L胰酶维持液,置37 ℃、5% CO2培养箱继续培养,分别于培养后24 h和48 h,取样进行相关指标检测。
3.3H9N2-SIV感染PMVEC后SOD、GSH-Px、ROS和NOS水平的测定 将细胞培养至对数生长期,使用不含EDTA的胰酶消化收集细胞,4 ℃ 800×g离心5 min,PBS洗涤,计数5×107个细胞,加0.8 mL缓冲液重悬,细胞悬液冰浴、超声波匀浆;加0.2 mL Medium Buffer混匀,4 ℃、10 000×g离心10 min,去上清;立即使用,进行蛋白定量,按照检测试盒说明书操作步骤测定并计算PMVEC中SOD、NOS、ROS及GSH-Px水平。
3.4H9N2-SIV感染PMVEC超微结构的观察 PMVEC培养至对数生长期,在细胞达到70%融合时,加入10-2稀释H9N2-SIV液500 μL进行感染,在感染后24 h和48 h,PBS缓冲液洗2次,用胰酶消化收集细胞,4 ℃、 800×g离心5 min,细胞沉淀经固定、脱水、包埋等处理后使用Leica EM UC7超薄切片机切片,切片经醋酸铀、枸橼酸铅染色,透射电镜(H-7650,120KV)观察。
3.5单层PMVEC跨膜电阻和通透性测定 参照刘亚楠等[10]报道的方法进行。根据单层PMVEC对辣根过氧化物酶(horseradish peroxidase, HRP)通透性的变化来反映单层PMVEC通透性的改变。即按105个/cm2细胞数将PMVEC接种于1%明胶包被的Transwell顶层小室微孔膜上,长至细胞融合后,将培养液更换为无血清无酚红DMEM培养液继续培养12 h,使细胞同步生长并处于静止期。用确定的最佳感染剂量的H9N2-SIV感染PMVEC,每个双层小室的顶室内加入3.4×10-6mol/L HRP,5%CO2、37 ℃孵育45 min;在病毒感染后24 h和48 h,采用WPIEVOM2电阻仪(WPI)测定Transwell小室上、下室之间的电阻,以此反映单层内皮细胞跨膜电阻值。同时,从每个双层小室底室内取2 μL样品,放于96孔板中,然后加入TMB显色液,使用多功能酶标仪在379 nm波长处读数,绘制标准曲线,以其占加入总HRP量的百分比(%)表示HRP浓度。
3.6H9N2-SIV感染PMVEC中TRPM2 mRNA的表达 收获PMVEC,将相同数量细胞移入离心管内1 600×g离心5 min去上清液, 倒置控干,按试剂盒说明书要求进行RNA提取,RNA浓度要求A260/A280大于2.0。根据Reverse Transcription System试剂盒说明书要求进行总RNA逆转录合成cDNA。TRPM2的上游引物序列为5’-GGCACCUUUAUAUCAUUAATT-3’,下游引物序列为5’-UUAAUGAUAUAAAGGUGCCTT-3’;GAPDH的上游引物序列为5’-GTGGAGTCATACTGGAACATGTAG-3’,下游引物序列为5’-AATGGTGAAGGTCGGTGTG-3’。加入上述引物,进行real-time PCR,条件:95 ℃ 10 s和55 ℃ 40 s为一个循环,共进行40个循环。在55 ℃条件下检测荧光信号,检测熔解曲线。用2-ΔΔCt法计算TRPM2 mRNA相对表达量。
3.7H9N2-SIV感染PMVEC中TRPM2蛋白的表达 收获细胞,在冰上进行裂解60 min;4 ℃、14 000×g离心5 min,取上清进行BCA蛋白定量。加入5×上样缓冲液,煮沸10 min,蛋白样品收集。将蛋白样品进行SDS-PAGE:条件为80 mA恒流4 h;将蛋白转移至PVDF膜上;TBST溶液配制的5%脱脂奶粉室温封闭60 min;抗TRPM2抗体 (1 ∶300释稀)和抗GAPDH抗体 (1 ∶1 000释稀) 4 ℃孵育过夜;TBS溶液洗2次,再用TBST溶液洗1次,每次10 min;II抗室温孵育1 h;TBS溶液洗2次,再用TBST溶液洗1次,每次10 min;ECL显色系统显影检测目的蛋白。
3.8H9N2-SIV感染PMVEC凋亡的观察 采用Annexin V/PI双染色法观察H9N2-SIV感染对PMVEC凋亡的影响。移出培养液,用胰蛋白酶消化成单个细胞,以预冷PBS润洗3次后,弃PBS液体,收集细胞。将收集的各组PMVEC悬浮于1×Annexin V/PI结合缓冲液中,细胞密度约为2×108/L,加入5 μL的Annexin V染色液和5 μL的PI染色液,轻轻混匀避光室温孵育15 min,立即在激光共聚焦显微镜(Olympus FV1200)下观察细胞凋亡情况。
4 统计学处理
用SPSS 16.0统计软件进行分析。数据均采用均数±标准差(mean±SD)表示,多组间比较采用单因素方差分析,组间两两比较采用最小显著性差异法(LSD法)。以P<0.05为差异有统计学意义。
结 果
1 H9N2-SIV感染PMVEC对NOS、ROS、GSH-Px和SOD水平的影响
在H9N2-SIV感染PMVEC后24 h开始,感染组中GSH-Px和SOD活性与未感染对照组相比开始出现显著下降(P<0.05);至感染后48 h,GSH-Px和SOD活性下降更为明显,与未感染的对照组相比有显著差异(P<0.01);与之相反,ROS水平和NOS活性则从感染后24 h开始显著高于未感染对照组(P<0.01),至感染后48 h,仍然显著高于对照组(P<0.01),见表1。
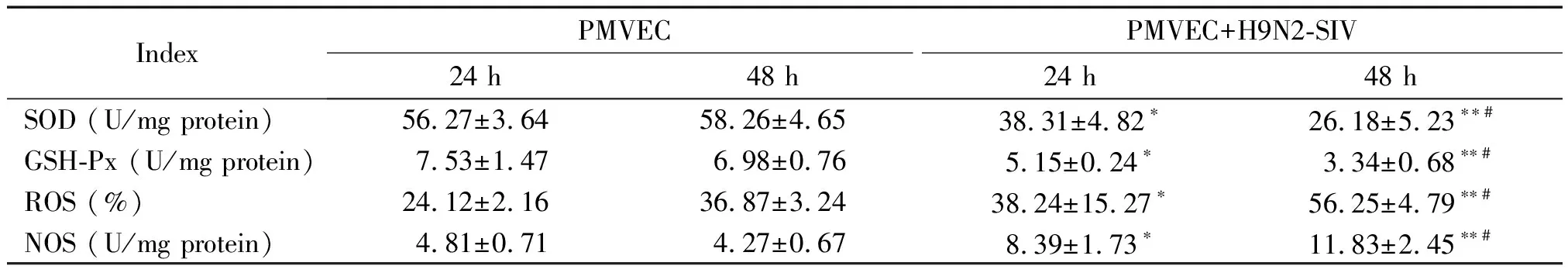
表1 PMVEC感染H9N2-SIV后SOD、GSH-Px、ROS和NOS的变化
2 H9N2-SIV感染导致PMVEC超微结构损伤
未加H9N2-SIV感染PMVEC电镜观察显示细胞形状结构完整,细胞质内见较多线粒体、内质网等细胞器结构,细胞核体积较大,双层核膜结构完整,染色质分布均匀,可见核仁,见图1A;然而,H9N2-SIV感染后24 h小鼠PMVEC可见胞浆中线粒体基质电子密度降低,嵴数量减少,部分区域呈空泡状,见图1B;细胞体积缩小,细胞器数量减少,在胞质内可见大小不等的多个空泡状结构,挤压细胞核至变形,可见核仁,见图1C;至感染后48 h,细胞核形状不规则,异染色质聚集在核膜下,细胞核电子密度降低,部分核膜断裂,有形成凋亡小体的趋势,见图1D。
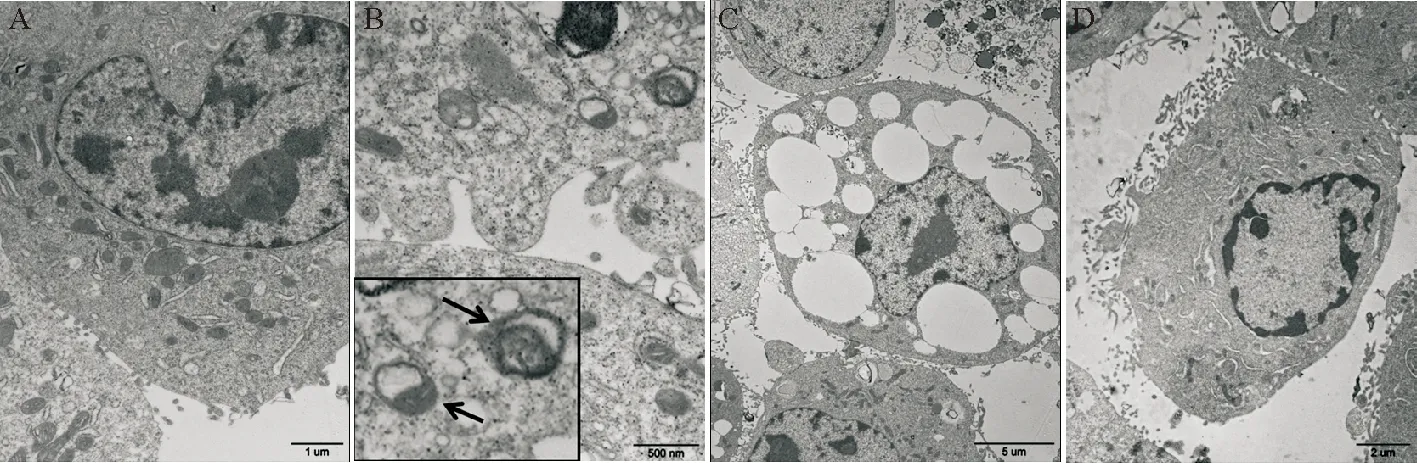
Figure 1.The ultrastructural changes of PMVEC infected by H9N2-SIV. A: representative normal control PMVEC without H9N2-SIV infection; B and C: representative PMVEC infected with H9N2-SIV at 24 h after infection; D: representative PMVEC infected with H9N2-SIV at 48 h after infection. The arrow showed mitochondrial damage.
3 H9N2-SIV感染对单层PMVEC跨膜电阻和通透性的影响
H9N2-SIV感染PMVEC后,随着病毒感染时间的延长,跨膜电阻值逐渐降低(P<0.05或P<0.01),小室内HRP含量增多,单层细胞通透性增高(P<0.05或P<0.01),见图2。

Figure 2.The effect of H9N2-SIV on PMVEC permeability. A: TEER; B: HRP influx. Mean±SD. n=3. *P<0.05, **P<0.01 vs PMVEC group.
4 H9N2-SIV感染显著增加PMVEC中TRPM2 mRNA表达
real-time PCR结果显示,H9N2-SIV感染PMVEC组TRPM2 mRNA表达量随着病毒感染时间延长而明显增加(P<0.01),见图3。
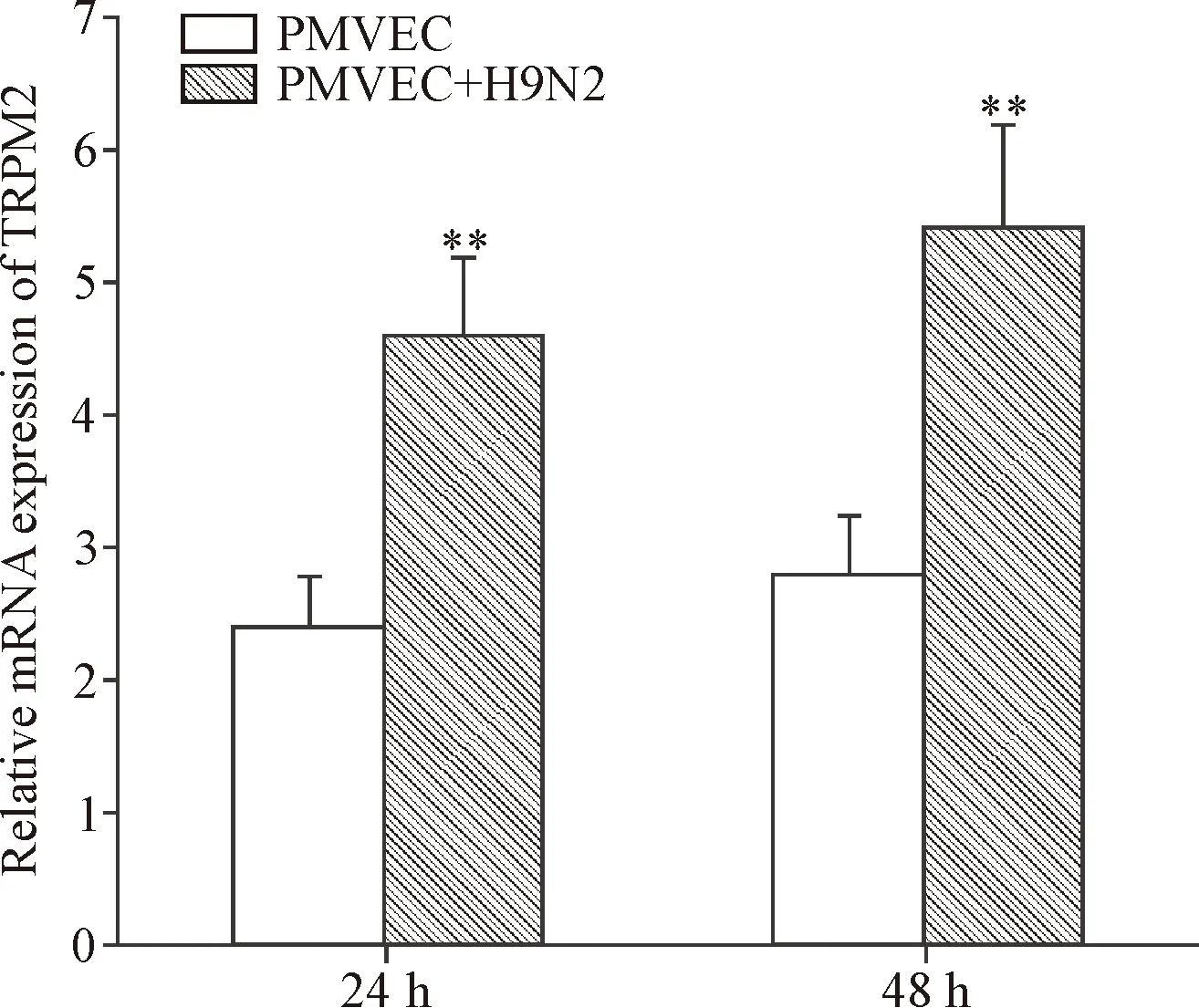
Figure 3.The mRNA expression of TRPM2 in PMVEC detected by real-time PCR. Mean±SD. n=3. **P<0.01 vs PMVEC group.
5 H9N2-SIV感染显著增加PMVEC中TRPM2蛋白表达
Westren blot检测结果显示,未感染PMVEC组在分子量171 kD出现明显的条带;H9N2-SIV感染PMVEC组24 h和48 h时均出现较未感染PMVEC组更明显的条带,显示病毒感染后TRPM2表达明显增加(P<0.01),见图4。
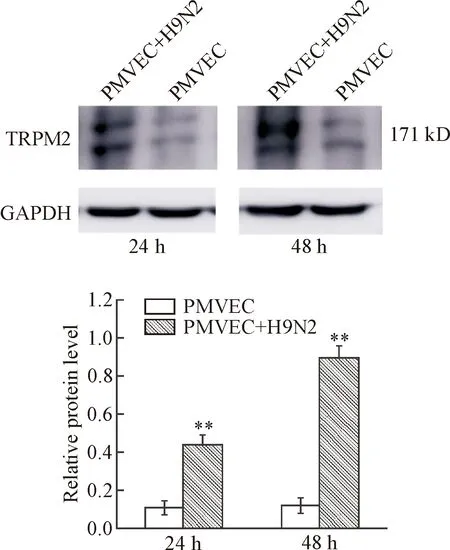
Figure 4.The protein expression of TRPM2 in H9N2-SIV-infected PMVEC detected by Western blot. Mean±SD. n=3. **P<0.01 vs PMVEC gorup.
6 H9N2-SIV感染对PMVEC凋亡的影响
H9N2-SIV感染PMVEC后,采用Annexin V/PI>双染色法激光共聚焦显微镜观察细胞凋亡情况。结果显示未经病毒感染的PMVEC对照组在培养24 h和48 h后视野内均未见明显的荧光;在H9N2-SIV感染PMVEC 24 h后,较多感染细胞细胞膜被染色呈现绿色荧光,其中部分细胞细胞核被染色呈现红色荧光;随着感染时间延长,至感染后48 h,更多细胞被染色呈现绿色和红色荧光,表明H9N2-SIV感染导致PMVEC出现明显的凋亡现象,见图5。
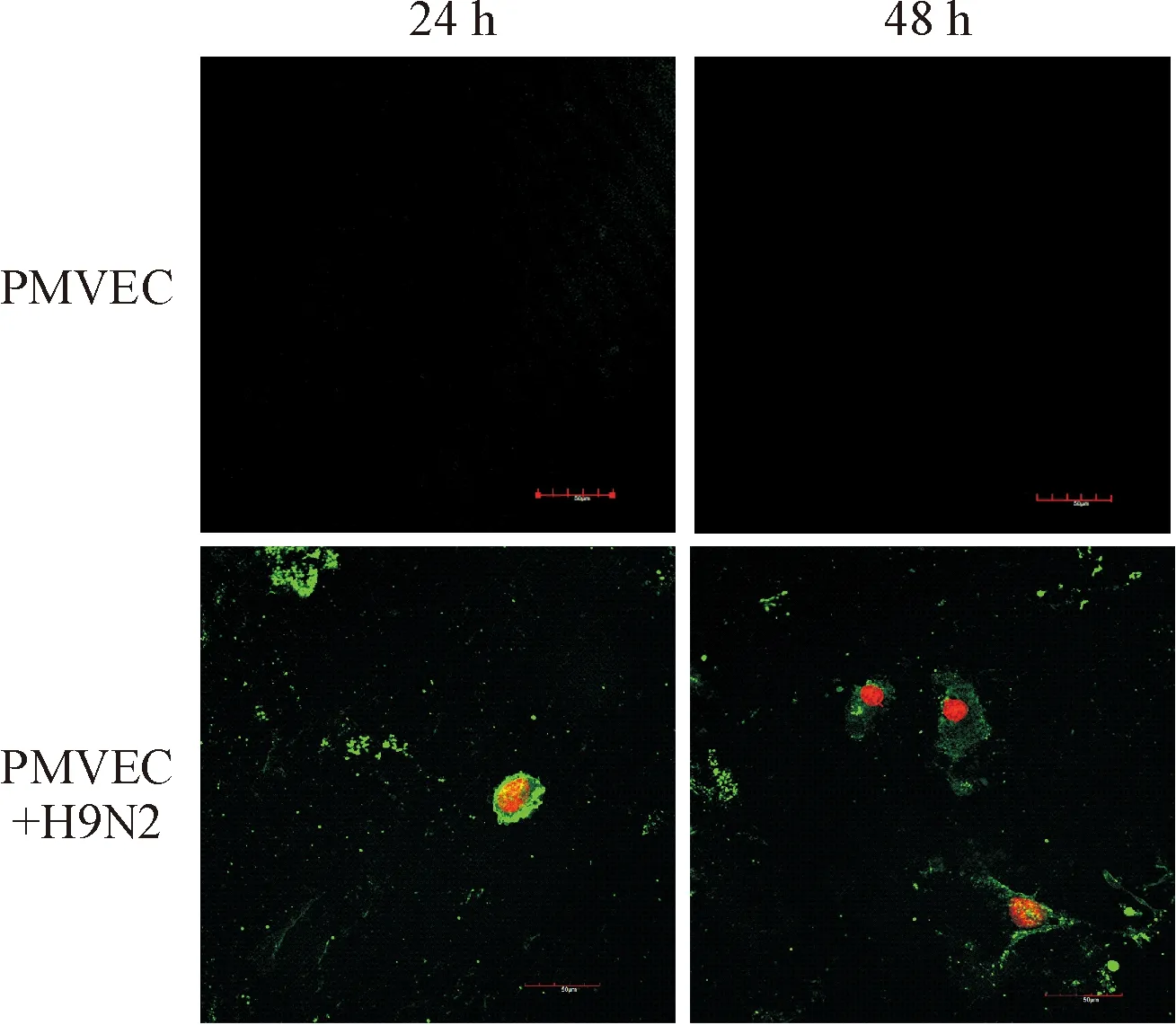
Figure 5.Apoptosis of PMVEC with H9N2-SIV infection under laser scanning confocal microscope (×400).
讨 论
自1997年首次发现H5N1禽流感病毒感染以来,不断出现新的不同亚型的动物源性流感病毒跨越种属屏障感染人,甚至引起较为严重的公共卫生事件,如H7N9、H5N2、H9N2以及H10N8等[11],表明动物源性流感病毒已逐渐成为威胁人类健康的重要病原体之一。在研究H5N1和H7N9等禽流感感染人类并引起严重肺损伤的机制时,有关炎症因子,尤其是氧化应激导致的血管内皮细胞损伤占有重要作用。
为了探讨流感病毒诱导血管内皮细胞损伤的作用机制,本研究利用H9N2-SIV感染小鼠肺微血管内皮细胞,结果显示病毒感染过程中ROS和NOS水平显著升高,但是SOD和GSH-Px活性则明显降低,表明H9N2-SIV感染由于ROS的大量产生以及因清除自由基消耗而导致SOD和GSH-Px明显降低。同时,电镜超微结构观察到病毒感染细胞过程中胞浆中细胞器数量减少,可见大量的空泡结构,挤压细胞核至变形;线粒体基质减少甚至消失。此外,细胞跨膜电阻显著下降以及通透性增加等均反映H9N2-SIV感染导致PMEVC出现明显的细胞损伤,这种损伤可能与大量ROS产生有关。He等[12]研究显示H5N1禽流感病毒感染小鼠肺损伤过程中诱导产生的活性氧类自由基发挥重要作用,流感病毒能够调节细胞内氧化还原敏感性信号通路促进病毒复制和致病作用。因此,探索抗氧化剂或干扰诱导产生自由基的相关信号通路的研究,成为目前干预流感病诱导肺损伤的研究热点之一[13]。
在研究氧化应激导致血管内皮细胞损伤的作用机制时,Hecquet等[4]发现H2O2诱导TRPM2表达显著升高并参与血管内皮细胞Ca2+内流,最终导致细胞通透性增高。Mayo等[14]在研究H5N1禽流感病毒致病机制时发现H5N1禽流感病毒可以诱导细胞外Ca2+内流增加,导致细胞凋亡发生和线粒体功能异常。此外,Sun等[15]也证实TRPM2在H2O2作用导致血管内皮细胞细胞凋亡中发挥作用。因此上述研究提示氧化应激体条件下诱导Ca2+内流增加以及线粒体损伤及细胞凋亡在致病机制中发挥了作用。TRPM2作为氧化应激敏感性非选择性钙离子通道,在氧化应激损伤诱导Ca2+内流和细胞凋亡等起着作用,然而,目前TRPM2在流感病毒感染致血管内皮细胞损伤的研究未见文献报道。因此,本实验中进一步观察H9N2-SIV感染过程中TRPM2 mRNA和蛋白的表达情况,结果显示H9N2病毒感染引起PMVEC内TRPM2 mRNA和蛋白表达均增加,提示TRPM2参与H9N2-SIV感染导致的PMVEC损伤过程。此外,有结果显示检测H9N2-SIV感染后Na+-K+-ATP显著下降(实验结果另文报道),同时Annexin V/PI双染色法激光共聚焦显微镜观察显示感染细胞细胞凋亡显著增多,以上结果证实H9N2-SIV感染PMVEC过程中线粒体活性显著下降,同时介导细胞凋亡增加。H2O2通过TRPM2活化Ca2+依赖性酪氨酸激酶Pyk2,引起Ca2+内流,导致Ras GTPase放大Erk信号通路,证实TRPM2是活性氧类自由基进一步引起炎症反应的重要级联信号通路之一[16]。在H2O2导致的氧化应激过程中抑制内源性TRPM2的表达和功能,从而干扰其介导的Ca2+内流可以减缓内皮细胞凋亡,有效缓解氧化应激导致的血管内皮细胞损伤[4, 17]。此外,TRPM2过表达的SH-SY5Y细胞在H2O2作用下细胞死亡显著增加[18]。以上研究表明TRPM2在氧化应激损伤致病机制中具有重要作用。本研究结果也提示TRPM2参与了流感病病毒在PMVEC的致病过程。这些研究为进一步通过干预TRPM2途径阐明流感病毒诱导肺损伤机制提供了实验依据。
猜你喜欢
临床医学工程(2022年3期)2022-04-20
中学生物学(2021年8期)2021-11-02
山西农业大学学报(自然科学版)(2020年1期)2020-03-04
祝您健康·文摘版(2019年2期)2019-06-11
科学24小时(2019年5期)2019-06-11
中国果业信息(2019年1期)2019-01-05
家庭百事通·健康一点通(2017年3期)2017-03-22
中国实用医药(2016年16期)2016-07-26
天津农业科学(2016年3期)2016-03-12
科学启蒙(2015年8期)2015-08-07
