
胰高血糖素样肽1通过Keap1-Nrf2信号通路减轻糖尿病大鼠心肌微血管损伤*
2019-12-25 06:10王东娟李恒栋谢小玲李立成
中国病理生理杂志 2019年12期
王东娟, 李恒栋, 谢小玲, 李立成
(中国科学院大学宁波华美医院心内科, 浙江 宁波 315010)
心血管并发症是导致糖尿病(diabetes mellitus,DM)患者死亡的主要原因之一,引起糖尿病心血管并发症的机制包括心肌间质纤维化、心肌能量代谢异常以及心肌微血管病变等[1]。已有研究证实,糖尿病心肌微血管损伤早于大血管及心肌细胞,与糖尿病心肌病发生发展密切相关,但是目前尚无有效的控制或逆转心肌微血管损伤的手段[2]。胰高血糖素样肽1(glucagan-like peptide-1, GLP-1)是由空、回肠上皮L细胞分泌的一种小分子多肽,研究表明除了降血糖作用外,GLP-1还具有抑制氧化应激反应、调节炎性因子释放和抗凋亡等多种生物学保护作用。
GLP-1的心血管保护作用受到了越来越多的关注,给予心衰大鼠GLP-1干预,可通过GLP-1受体(GLP-1 receptor, GLP-1R)介导下游信号通路抑制心室重塑[3];在缺血再灌注大鼠模型,给予GLP-1处理可通过抑制氧化应激反应减轻缺血再灌注损伤[4],提示GLP-1发挥心血管保护作用的重要机制之一即抗氧化应激作用。氧化应激损伤是糖尿病多种并发症的统一机制。Kelch样环氧氯丙烷相关蛋白1(Kelch-like epichlorohydrin-associated protein 1, Keap1)-核因子E2相关因子2(nuclear factor E2-related factor 2, Nrf2)信号通路是机体重要的抗氧化应激信号通路,可通过调节下游信号分子发挥抗氧化应激效应,激活Keap1-Nrf2信号通路对多种糖尿病相关并发症有益,如糖尿病肾病、糖尿病神经病变以及糖尿病视网膜病变等[5]。但是GLP-1调控Keap1-Nrf2信号通路发挥糖尿病心肌微血管的保护作用尚无文献报道。因此,本项工作建立糖尿病大鼠模型,拟通过体内和体外实验,明确GLP-1对糖尿病大鼠心肌微血管的保护作用,并进一步探讨其保护作用是否通过Keap1-Nrf2信号通路介导。
材 料 和 方 法
1 动物
清洁级雄性6~8周龄Sprague-Dawley(SD)大鼠40只,体重160~220 g,18只用于动物实验,22只用于心肌微血管内皮细胞原代分离培养。由宁波大学实验动物中心提供,许可证号SYXK(浙)2019-0005。
2 主要试剂
链脲佐菌素(streptozocin, STZ)购自Sigma;GLP-1和GLP-1类似物艾塞那肽(exenatide)购自Tocris;DMEM培养液和胎牛血清购自HyClone;体外血管通透性测定试剂盒购自Chemicon;超氧化物检测试剂盒和超氧化物阴离子荧光探针二氢乙啶(dihydroethidium, DHE)购自碧云天生物技术公司;GLP-1R抗体和Nrf2 siRNA购自Santa Cruz;抗Keap1、Nrf2、血红素加氧酶-1(heme oxygenase-1, HO-1)和β-actin抗体购自Abcam;BCA蛋白定量试剂盒购自Pierce;其它试剂均为进口或国产分析纯。
3 主要方法
3.1糖尿病大鼠模型构建[6]将STZ溶于0.1 mmol/L柠檬酸缓冲液中,配制成0.25%的STZ注射液。大鼠腹腔注射STZ注射液(35 mg·kg-1·d-1),连续3 d;尾静脉取血测量血糖,若3次随机血糖≥16.7 mmol/L,认为糖尿病大鼠建模成功。将糖尿病大鼠随机分为2组:DM组(n=6),予以糖尿病大鼠腹腔注射生理盐水;DM+exenatide组(n=6),予以糖尿病大鼠腹腔注射艾塞那肽0.25 μg/kg,每天2次,连续干预3个月。以年龄、性别匹配的正常SD大鼠作为对照组(n=6),干预前后检测各组大鼠体重、血糖、血压等变化。
3.2透射电镜检测心肌微血管通透性 硝酸镧是直径约4 nm的重金属颗粒,生理状态下不可穿越细胞连接,因此可通过硝酸镧灌注法评估心肌微血管通透性变化。采用改良Krebs-Henseleit溶液配制二甲砷酸钠缓冲液(0.2 mol/L),再利用二甲砷酸钠缓冲液配置2%硝酸镧灌流液[6]。采用Langendorff装置,取硝酸镧灌流液以生理压力灌流各组大鼠心脏20 min。取左心室组织,常规戊二醛固定、包埋、切片,透射电镜下通过观察硝酸镧的积聚部位评估心肌微血管通透性变化。
3.3心肌微血管内皮细胞分离培养 0.2%戊巴比妥钠腹腔注射麻醉大鼠,无菌条件下取左室,以75%乙醇浸泡去除心内外膜,然后将心肌组织剪成小组织块。0.2% Ⅱ型胶原酶,37 ℃水浴消化5 min;0.25%胰酶,37℃水浴消化5 min;然后加入含10%胎牛血清的DMEM培养液终止消化。室温下230 r/min离心10 min,弃上清,将细胞重悬于含15% 胎牛血清的DMEM培养液。培养6 h后换液去除未贴壁细胞,24 h后再次换液,以后每3 d换液1次。细胞生长至80%融合后消化传代,第2~3代细胞用于进一步实验[7]。心肌微血管内皮细胞分为以下3组[6]:正常对照(control)组,培养液葡萄糖浓度为5.5 mmol/L; 高糖(high glucose,HG)组,培养液葡萄糖浓度为25 mmol/L;GLP-1处理组(HG+GLP-1组),培养液葡萄糖浓度为25 mmol/L,予以GLP-1(10-8mol/L)预处理。按照说明书,以si-Nrf2转染心肌微血管内皮细胞抑制Nrf2表达,以scrambled siRNA(NC-siRNA)作为阴性对照。
3.4单层心肌微血管内皮细胞通透性检测 取生长状态良好的心肌微血管内皮细胞,以每孔2×105的浓度接种于体外血管通透性测定试剂盒。置于37℃、5% CO2培养箱中,待细胞生长融合至80%左右时,将通透性物质FITC-dextran(FITC标记的葡聚糖,终浓度1 g/L)加入试剂盒内池,室温放置10 min,收集外池培养液,采用分光光度仪检测各组培养液荧光强度,并分析各组内皮细胞通透性变化。
3.5活性氧(reactive oxygen species, ROS)检测 采用超氧化物检测试剂盒检测ROS生成。取第2~3代心肌微血管内皮细胞接种于96孔板,待细胞生长融合至80%左右时,吸去培养液,PBS冲洗1次。每孔加入200 μL超氧化物检测工作液,37 ℃孵育3 min;按照预先分组处理各组细胞后,在450 nm测定吸光度(A值)。
3.6超氧化物阴离子检测 采用DHE法检测超氧化物阴离子生成。取第2~3代心肌微血管内皮细胞接种于盖玻片,待细胞贴壁后,按照预先分组处理各组细胞。以二甲基亚砜(dimethyl sulfoxide, DMSO)溶解DHE,然后将DHE溶液加入细胞培养液中(终浓度为5 μmol/L),孵育约30 min。将细胞爬片取出,PBS冲洗后,DAPI染核。激光共聚焦显微镜(激发波长535 nm)下观察各组细胞超氧化物阴离子变化。
3.7Western blot 各组细胞按照预先分组处理后,分别提取核蛋白和总蛋白;采用BCA蛋白定量试剂盒测定各组蛋白含量。配制SDS-PAGE聚丙烯酰胺凝胶,蛋白(20 μL)上样、电泳、转膜、脱脂牛奶封闭。加入I抗(GLP-1R、Keap1、Nrf2、HO-1和β-actin)4℃孵育过夜;洗膜,加入辣根过氧化物酶标记的II抗,室温孵育1 h。化学发光试剂滴膜,凝胶图像分析系统扫描,测定条带积分灰度值。
4 统计学处理
实验数据以均数±标准差(mean±SD)表示,采用SPSS 21.0统计软件进行单因素方差分析(one-way ANOVA)或t检验。以P<0.05为差异有统计学意义。
结 果
1 各组大鼠一般情况比较
糖尿病大鼠建模成功后,给予生理盐水或艾塞那肽干预3个月。干预前后检测各组大鼠体重、血糖和血压等变化。结果显示,与正常对照组相比,糖尿病组(生理盐水干预)大鼠血糖显著升高(P<0.05),体重显著降低(P<0.05),血压无显著变化;予以小剂量艾塞那肽干预后,与生理盐水干预组相比,体重、血压和血糖水平无显著性变化,见表1。

表1 各组大鼠一般情况比较
2 艾塞那肽改善糖尿病大鼠心肌微血管通透性
正常对照组大鼠心肌微血管内皮细胞连接完整,未见硝酸镧颗粒穿透基底膜;在糖尿病组大鼠,可见硝酸镧颗粒透过内皮细胞侵入基底膜及周围组织;而给予艾塞那肽干预后,可见大部分硝酸镧颗粒位于管腔和管壁,少部分位于基底膜,见图1。

Figure 1.Effects of exenatide on cardiac microvascular permeability observed under transmission electron microscope. The scale bar=1 mm. Lanthanum nitrate is highlighted with yellow arrows.
3 心肌微血管内皮细胞分离培养
心肌微血管内皮细胞采用酶消化法体外分离培养,培养至第3天时,细胞趋于伸展,呈多角形、近圆形或梭型,见图2A;培养至第7天时,呈典型“铺路石样”生长,见图2B。采用Western blot法检测观察到心肌微血管内皮细胞表达GLP-1R,见图2C。
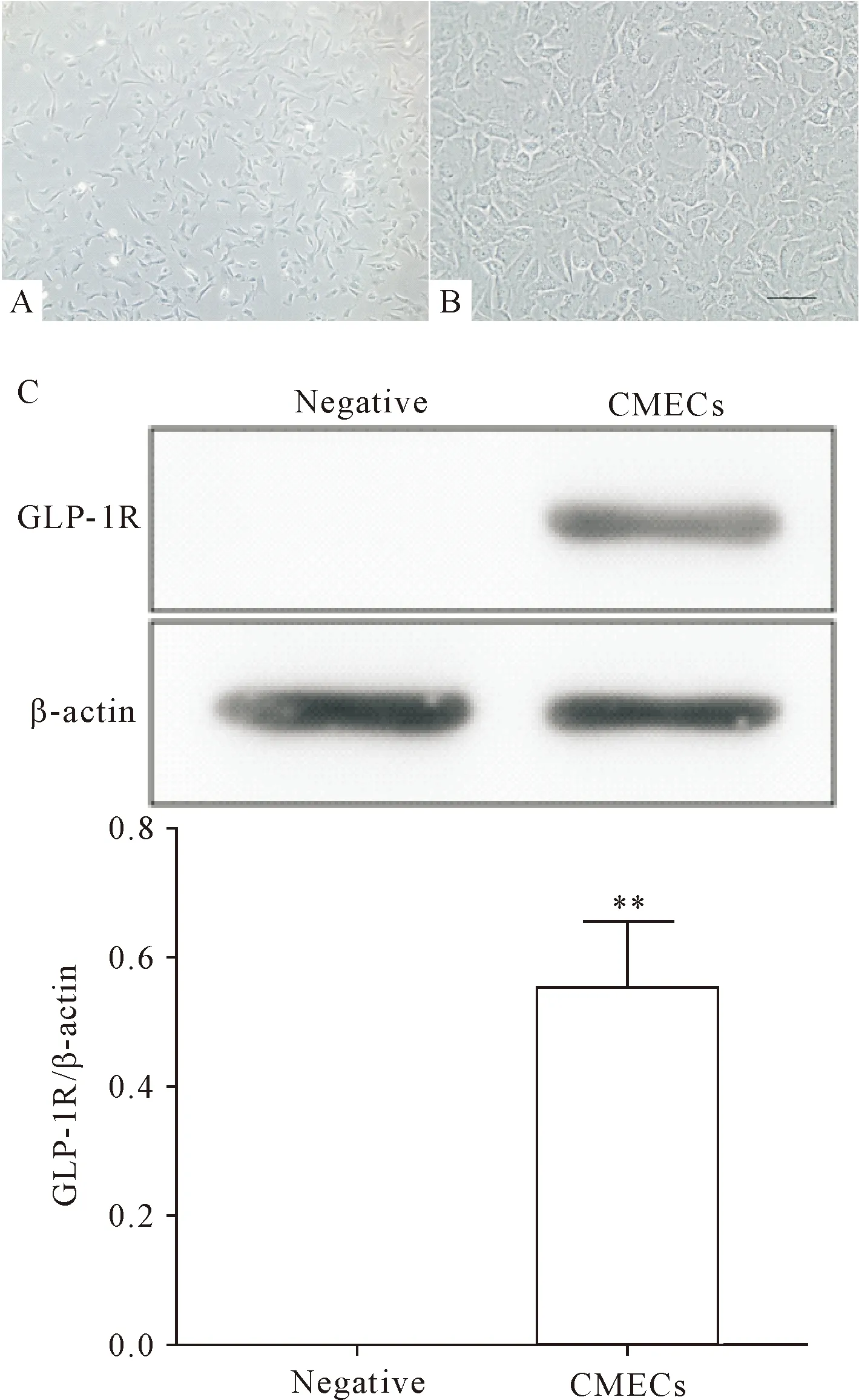
Figure 2.Characterization of cardiac microvascular endothelial cells (CMECs) and detection of GLP-1R expression. A: CMECs cultured for 3 d. B: CMECs cultured for 7 d. The scale bar=40 μm.C: GLP-1R expression detected by Western blot. Mean±SD. n=4.**P<0.01 vs negative group.
4 GLP-1改善高糖诱导的心肌微血管内皮细胞通透性损伤
与对照组相比,高糖作用下FITC-dextran渗透显著增多(P<0.01),而GLP-1干预后FITC-dextran渗透显著减少(P<0.05),见图3。

Figure 3.Effects of GLP-1 on high glucose (HG)-induced permeability of cardiac microvascular endothelial cells.Mean±SD. n=5.**P<0.01 vs control group; #P<0.05 vs HG+vehicle group.
5 GLP-1抑制高糖诱导的心肌微血管内皮细胞ROS生成
通过DHE染色观察到,与对照组比较,高糖处理组心肌微血管内皮细胞荧光强度显著增强(P<0.01),而给予GLP-1预处理后,心肌微血管内皮细胞荧光强度显著减弱(P<0.05),见图4A。ROS试剂盒检测进一步证实,高糖作用下心肌微血管内皮细胞生成ROS增多(P<0.01),而给予GLP-1预处理后心肌微血管内皮细胞生成ROS显著降低(P<0.05),见图4B。

Figure 4.Effects of GLP-1 on high glucose (HG)-induced oxidative stress in cardiac microvascular endothelial cells. A: the results of DHE staining (red, DHE; blue, DAPI; the scale bar=25 μm); B: ROS production detected by superoxide assay kit.Mean±SD. n=5.**P<0.01 vs control group; #P<0.05 vs HG+vehicle group.
6 GLP-1对高糖诱导的心肌微血管内皮细胞Keap1、Nrf2和HO-1表达的影响
心肌微血管内皮细胞在高糖作用下,keap1蛋白表达显著升高(P<0.01),核蛋白Nrf2表达显著减低(P<0.01),抗氧化应激相关分子HO-1显著降低(P<0.01);而给予GLP-1预处理的心肌微血管内皮细胞,与单纯高糖组相比,Keap1表达降低(P<0.05),Nrf2和HO-1表达显著升高(P<0.05),见图5。
7 GLP-1通过Keap1-Nrf2信号通路抑制高糖诱导的心肌微血管内皮细胞ROS生成
高糖作用下心肌微血管内皮细胞ROS生成增多(P<0.01),给予GLP-1预处理后ROS 生成降低(P<0.05),而预先予以si-Nrf2处理可抵消GLP-1对高糖诱导的心肌微血管内皮细胞ROS生成的抑制作用(P<0.05),给予NC-siRNA处理ROS生成无显著性变化(P>0.05),见图6。
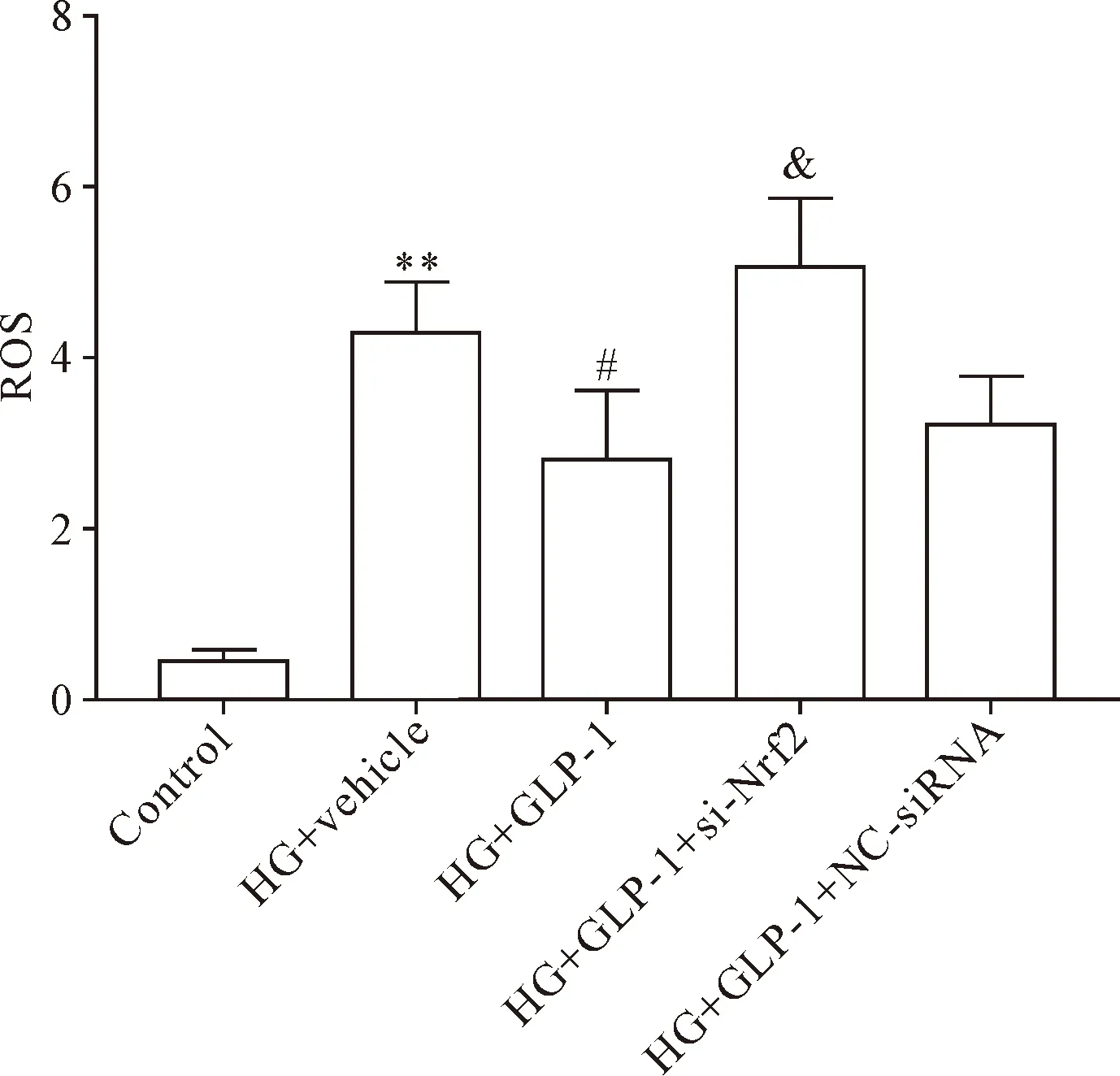
Figure 6.GLP-1 inhibited HG-induced ROS production in car-diac microvascular endothelial cells through Keap1-Nrf2 pathway.Mean±SD.n=5.**P<0.01 vs control group; #P<0.05 vs HG+vehicle group; &P<0.05 vs HG+GLP-1 group.
讨 论
心肌微血管内皮细胞在心肌中数量最多,糖尿病心肌微血管内皮细胞损伤早于大血管及心肌细胞,可进一步引起心脏微循环灌注不良、心肌能量代谢异常及心肌间质纤维化[8]。目前认为引发糖尿病心肌微血管损伤的可能机制包括氧化应激反应增强、蛋白激酶C活化、晚期糖基化终产物生成增加和多元醇通路激活等。但是,糖尿病心肌微血管病变的具体机制仍未完全阐明,目前仍无有效的控制心肌微血管病变的措施。
有科学家提出“氧化应激反应增强是糖尿病并发症的统一机制”[9-10]。在高脂喂养的糖尿病ApoE-/-小鼠,阿托伐他汀可通过类视黄醇X受体α(retinoid X recptor α, RXRα)抑制氧化应激反应,进一步抑制动脉粥样斑块形成[11]。在STZ诱导的糖尿病大鼠,予以运动(跑台)干预后,心肌组织NADPH氧化酶亚基p47phox和gp91phox表达降低[12],以上研究提示,抑制氧化应激反应对于保护糖尿病心血管并发症具有潜在意义。目前实验研究常用的抗氧化剂有维生素C、维生素E、α-硫辛酸、超氧化物歧化酶、褪黑素等,但是其研究结果并不令人十分满意。
GLP-1是具有多器官保护作用的胃肠道内分泌激素,近年研究提示其对内皮细胞、心肌细胞等也有保护功能[13]。GLP-1在体内可迅速被二肽基肽酶4降解而失去活性,因此在体一般给予GLP-1激动剂或二肽基肽酶4抑制剂。缺血前或再灌注同时给予离体或在体大鼠心脏输注GLP-1,可降低心肌梗死面积,但是予以GLP-1受体拮抗剂(9-39)干预后其保护作用消失[14]。给予糖尿病大鼠皮下注射利拉鲁肽(二肽基肽酶4抑制剂),研究显示利拉鲁肽并没有影响血浆血糖、胰岛素水平以及体重,但是在利拉鲁肽治疗的糖尿病大鼠心肌组织PKC表达水平降低,氧化应激反应下降,心肌脂肪变性改善,其作用机制可能是通过AMPK-Sirt1信号通路介导的[15]。但是,GLP-1对糖尿病心肌微血管保护作用的研究仍较少,其发挥保护作用的机制尚未完全阐明,有待于更加深入的探讨。本研究中,我们采用的是STZ腹腔注射诱导1型糖尿病模型,该模型排除了肥胖和胰岛素抵抗等因素的干扰,有助于明确GLP-1对高糖诱导的心肌微血管的保护作用。研究结果提示GLP-1激动剂艾赛那肽可改善糖尿病大鼠心肌微血管屏障功能。此外,小剂量的艾赛那肽并未显著降低糖尿病大鼠的血糖,从一定程度上排除了GLP-1的降糖作用所致的心血管保护作用。进一步的体外实验观察到GLP-1干预可显著抑制高糖诱导的心肌微血管内皮细胞氧化应激反应,并改善单层心肌微血管内皮细胞屏障功能,提示GLP-1发挥保护作用的机制可能是通过抑制氧化应激反应实现的。
Keap1-Nrf2信号通路是机体重要的抗氧化信号通路,Keap1-Nrf2信号通路失活可导致活性氧自由基在体内的积累[16]。糖尿病状态下激活 Keap1-Nrf2信号通路可显著减少ROS生成,并改善由氧化应激诱导出现的组织细胞功能紊乱。Sitagliptin可调节糖尿病肾脏的氧化应激反应,其机制可能是通过下调miR-200a,进一步激活Keap1-Nrf2信号通路实现的[17]。引起糖尿病视网膜病变的一个主要原因也是氧化应激损伤,通过激活Nrf2通路,可以抑制氧化应激,延缓糖尿病视网膜病变的发生发展[18]。硫化氢可缓解STZ诱导的糖尿病小鼠动脉粥样硬化进程,机制可能是激活Keap1-Nrf2信号通路抑制氧化应激反应[19]。Keap1-Nrf2信号通路可能是防治糖尿病相关并发症的一个有前景的潜在靶点,但是在糖尿病心肌微血管损伤领域目前仍缺少相关研究。在本研究中,高糖作用下心肌微血管内皮细胞Keap1蛋白表达升高,Nrf2和HO-1蛋白表达降低,而给予GLP-1干预后,Nrf2和HO-1蛋白表达升高,Keap1蛋白表达减低,提示Keap1-Nrf2信号通路可能参与GLP-1对高糖诱导的心肌微血管内皮细胞的保护作用;进一步采用si-Nrf2转染心肌微血管内皮细胞,si-Nrf2处理可抵消GLP-1对高糖诱导的心肌微血管内皮细胞的保护作用。
综上所述,GLP-1可抑制高糖诱导的大鼠心肌微血管内皮细胞氧化应激反应,改善心肌微血管屏障功能,这些保护效应可能是通过Keap1-Nrf2信号通路介导实现的。
猜你喜欢
眼科新进展(2022年12期)2022-12-29
体育科技文献通报(2022年4期)2022-10-21
世界科学技术-中医药现代化(2022年3期)2022-08-22
现代仪器与医疗(2022年2期)2022-08-11
眼科新进展(2022年2期)2022-03-11
祝您健康·文摘版(2021年12期)2021-12-08
现代临床医学(2021年5期)2021-11-02
天津医科大学学报(2021年4期)2021-08-21
昆明医科大学学报(2021年3期)2021-07-22
世界最新医学信息文摘(2021年12期)2021-06-09
