
超高效液相色谱-四极杆/静电场轨道阱高分辨质谱快速筛查鱼和虾样品中200种药物残留
2019-03-28 09:52于慧娟娄晓祎汤云瑜
质谱学报 2019年2期
于慧娟,汪 洋,2,孔 聪,娄晓祎,韩 峰,王 媛,汤云瑜
(1.东海水产研究所,农业部水产品风险评估实验室(上海),上海 200090;2.上海海洋大学食品学院,上海 201306)
水产品中的药物残留主要来源于养殖过程中渔药和饲料的使用。目前,存在向渔药和饲料中非法添加药物的现象,质量良莠不齐[1-3]。将质量不合格的渔药、饲料用于水产动物养殖过程,会导致水产品中的药物残留呈现杂乱未知状态,难以预测其中的安全隐患。因此,建立高效可靠的水产品中多种药物同时快速筛查的方法,对水产品中药物残留风险监测和隐患排查具有十分重要的意义。
目前,食品中药物残留的检测方法主要为液相色谱-三重四极杆串联质谱法[4-6],并通过多反应监测模式或选择反应监测模式实现多种化合物的同时定性与定量[7-8]。由于三重四极杆质谱仪的分辨率和质量精度较低,该方法局限于检测结构类似的同族化合物或少数其他几类化合物[9-10]。
随着兽药的商业化,越来越多的化合物种类需要监测[11]。高分辨质谱凭借高分辨率和高质量精度为多残留检测提供了新的手段,例如飞行时间质谱[12-14]和静电场轨道阱质谱[15-21],这些技术不仅能同时分析上百种化合物,还可以进行非目标物的筛查。采用液相色谱-四极杆/静电场轨道阱高分辨质谱法快速筛查水产品中药物残留是近几年新兴的一种检测手段,相关研究报道较少。杨璐齐等[20]和Sherri等[21]运用此技术分别同时检测了水产品中33种、260种药物,虽然Sherri等测定了260种药物,但经实际样品验证的药物仅为70多种。
本研究拟建立超高效液相色谱-四极杆/静电场轨道阱质谱法,通过优化分析条件,开发快速、高通量的样品前处理方法,实现水产品中200种药物残留的同时快速检测,并通过建立化合物数据库对目标物进行定性。希望为快速了解水产品中药物残留状况、隐患因子排查提供有效的技术支撑。
1 实验部分
1.1 主要仪器与装置
Ultimate 3000 超高效液相色谱-Q-Exactive静电场轨道阱高分辨质谱联用系统:配有TraceFinder分析软件,美国Thermo Fisher公司产品;0.22 μm尼龙水相针式滤膜:美国Supelco公司产品;涡旋搅拌器:德国IKA公司产品;16RXII高速冷冻离心机:日本 HITACHI CF公司产品;Milli-Q超纯水机:美国Millipore公司产品;PL602-L电子天平:瑞士Mettler-Toledo公司产品;BT 125D精密天平:德国Sartorius公司产品;KQ-300E超声波提取仪:昆山市超声仪器有限公司产品。
1.2 主要材料与试剂
200种风险排查标准品:均为德国Dr. Ehrenstofer GmbH公司产品;乙腈、甲醇、乙酸乙酯和甲酸:均为色谱纯,美国J.T.Baker公司产品;实验用水:由Milli-Q超纯水机制备;Oasis PRiME HLB固相萃取柱(200 mg/3 mL):美国Waters公司产品。
1.3 实验条件
1.3.1样品制备 从农贸市场购买草鱼、鲫鱼、鳊鱼和南美白对虾等鲜活水产品。取鱼可食用部分,均质混匀;将虾去头、去壳,均质混匀;分别装入聚乙烯瓶中,于-18 ℃冷冻保存,测定前解冻,称样。
1.3.2样品前处理 准确称取2.00 g样品于30 mL聚丙烯离心管中,加入200 μL 0.1 mol/L EDTA-Na2和10 mL乙腈,在高通量研磨仪中,以2 000 r/min振荡30 s,超声10 min,涡旋2 min,于5 ℃以10 000 r/min离心10 min,取出上清液。向残渣中加入10 mL乙酸乙酯,按上述方法重复操作1次,合并2次上清液,于40 ℃减压蒸发至近干,用3 mL乙腈溶解,以6 000 r/min离心10 min,得到待净化液。
将待净化液加入SPE固相萃取柱中,用1 mL乙腈淋洗,接收全部上样流出液至10 mL离心管中,40 ℃氮气吹至近干,用1 mL含0.1%甲酸的乙腈-水溶液(40∶60,V/V)复溶,于5 ℃以10 000 r/min离心10 min,取上清液,过0.22 μm尼龙针式滤膜,待测。
1.3.3色谱条件 色谱柱:Thermo Accuore aQ柱(100 mm×2.1 mm×2.6 μm);流动相:0.1%甲酸水溶液(A)和0.1%甲酸-乙腈溶液(B);梯度洗脱程序:0~1.0 min(100%A),1.0~18.0 min(100%~0%A),18.0~22.0 min(100%A);流速:0.3 mL/min;柱温:30 ℃;进样量:10 μL。
1.3.4质谱条件 静电场轨道阱质谱;可加热的电喷雾离子源(HESI-Ⅱ);喷雾电压:3.2 kV(+),2.8 kV(-);鞘气:氮气,流速8 L/min;辅助气:氮气,流速10 L/min,温度350 ℃;吹扫气:氮气,流速1.5 L/min;离子传输管温度:325 ℃;质谱数据获取模式:一级母离子全扫描加数据依赖的二级子离子扫描模式(Full MS/dd-MS2);Full MS分辨率:70 000;最大注入时间:100 ms;质量扫描范围:m/z150~1 000;二级质谱分辨率:17 500;触发阈值:2×105;最大注入时间80 ms。200种药物的质谱参数列于表1。
1.3.5标准溶液的配制 准确称取适量的标准品,用甲醇配制成约500 mg/L的单标储备液,不溶药品加入1 mL甲酸或二甲亚砜溶解后,用甲醇定容,于-42 ℃避光保存。将200种药物按性质分为18类,即苯并咪唑类、β-受体激动剂类、β-内酰胺类、氯霉素类、染料类、激素类、大环内酯类、硝基呋喃类、喹诺酮类、喹噁啉类、镇静剂类、磺胺类、三嗪类、四环素类、硝唑类、咪唑类农药、有机氯类农药和有机磷类农药,分别吸取适量的单标储备液,用甲醇配制成18种10 mg/L混合标准溶液;再分别吸取适量的18种混合标准溶液,用甲醇稀释,配制成浓度均为0.5 mg/L的200种药物混合标准工作液,于-42 ℃避光保存。
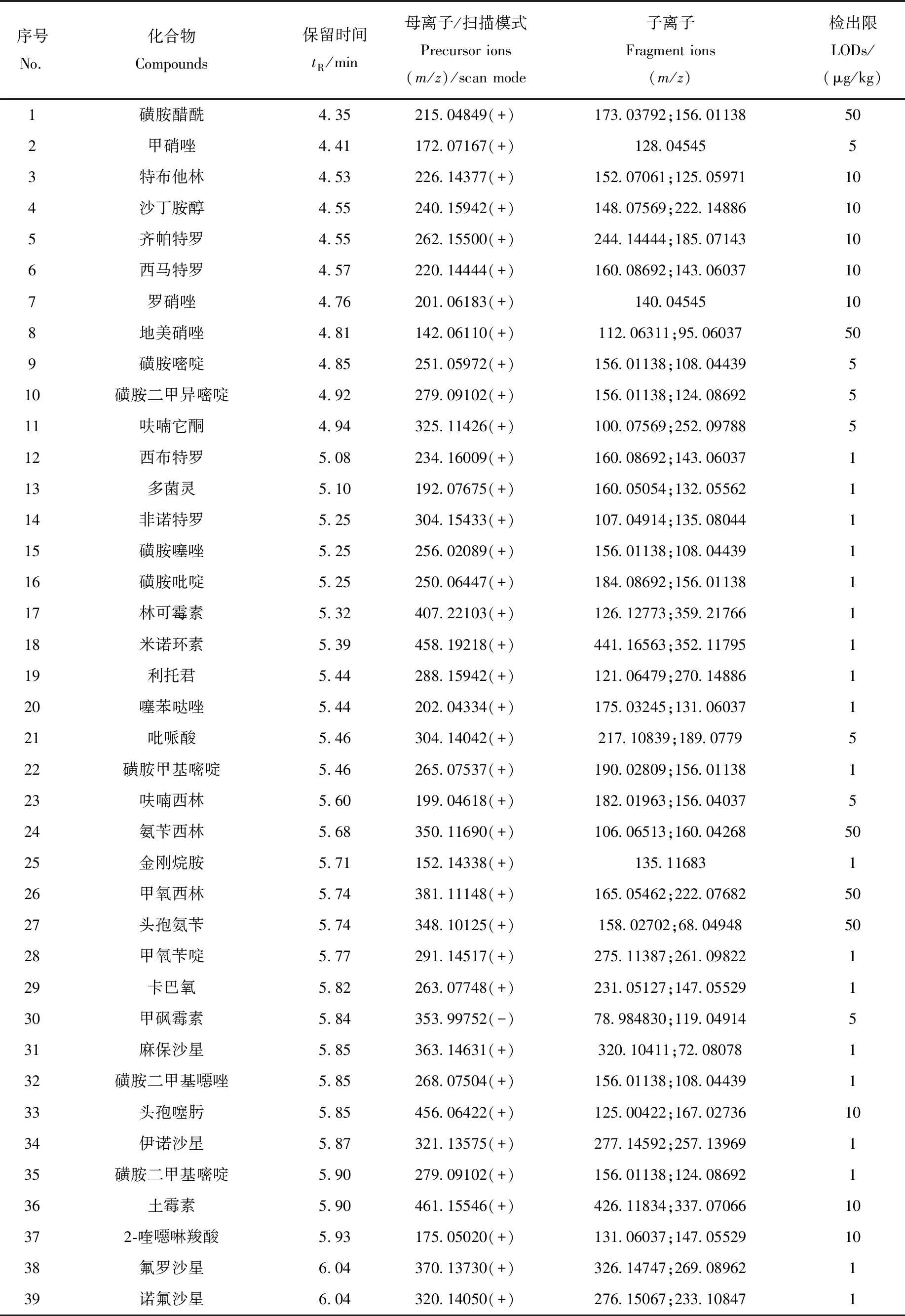
表1 200种化合物的色谱、质谱信息和检出限Table 1 Chromatographic and mass spectrometric parameters of 200 compounds

续表1

续表1
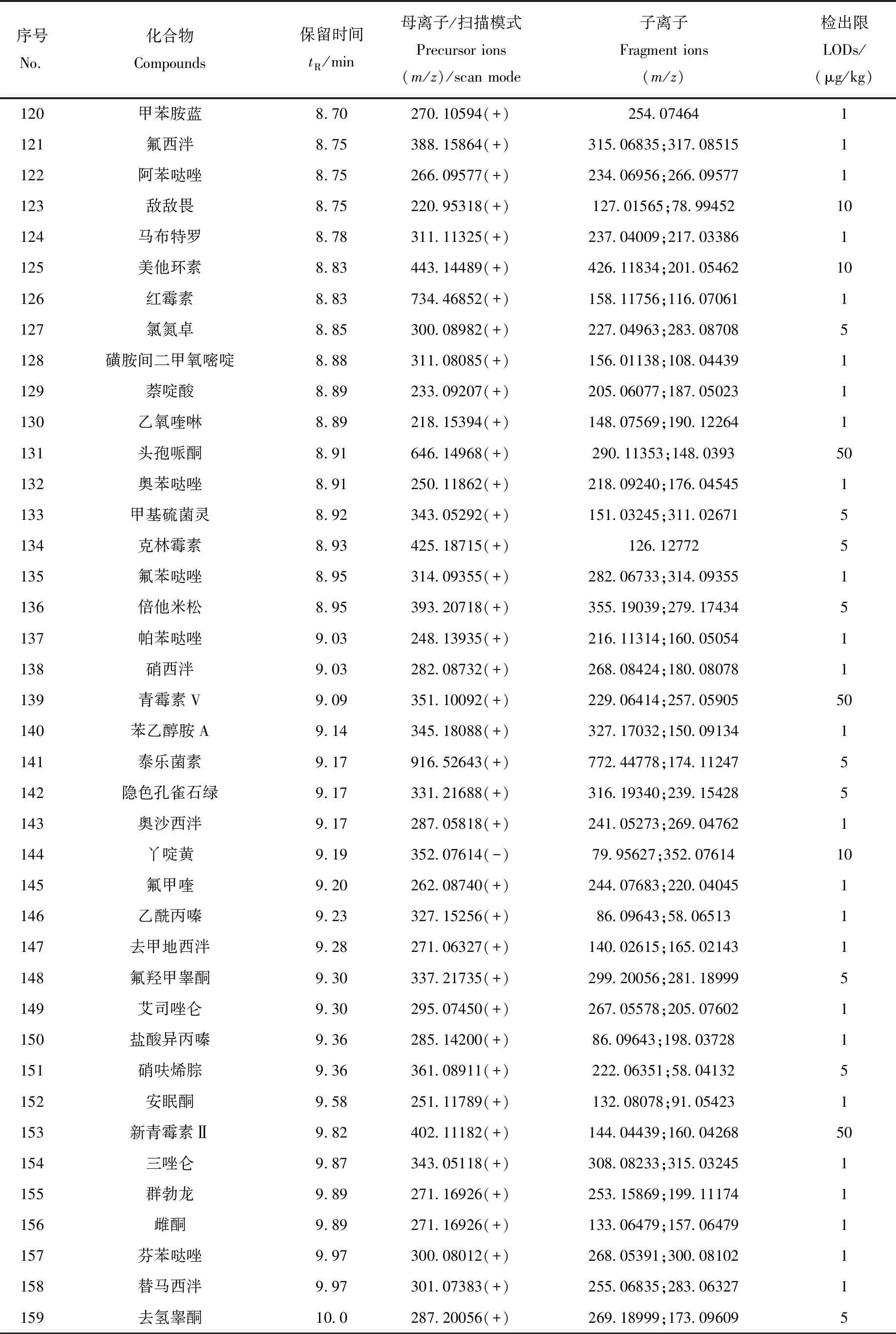
续表1
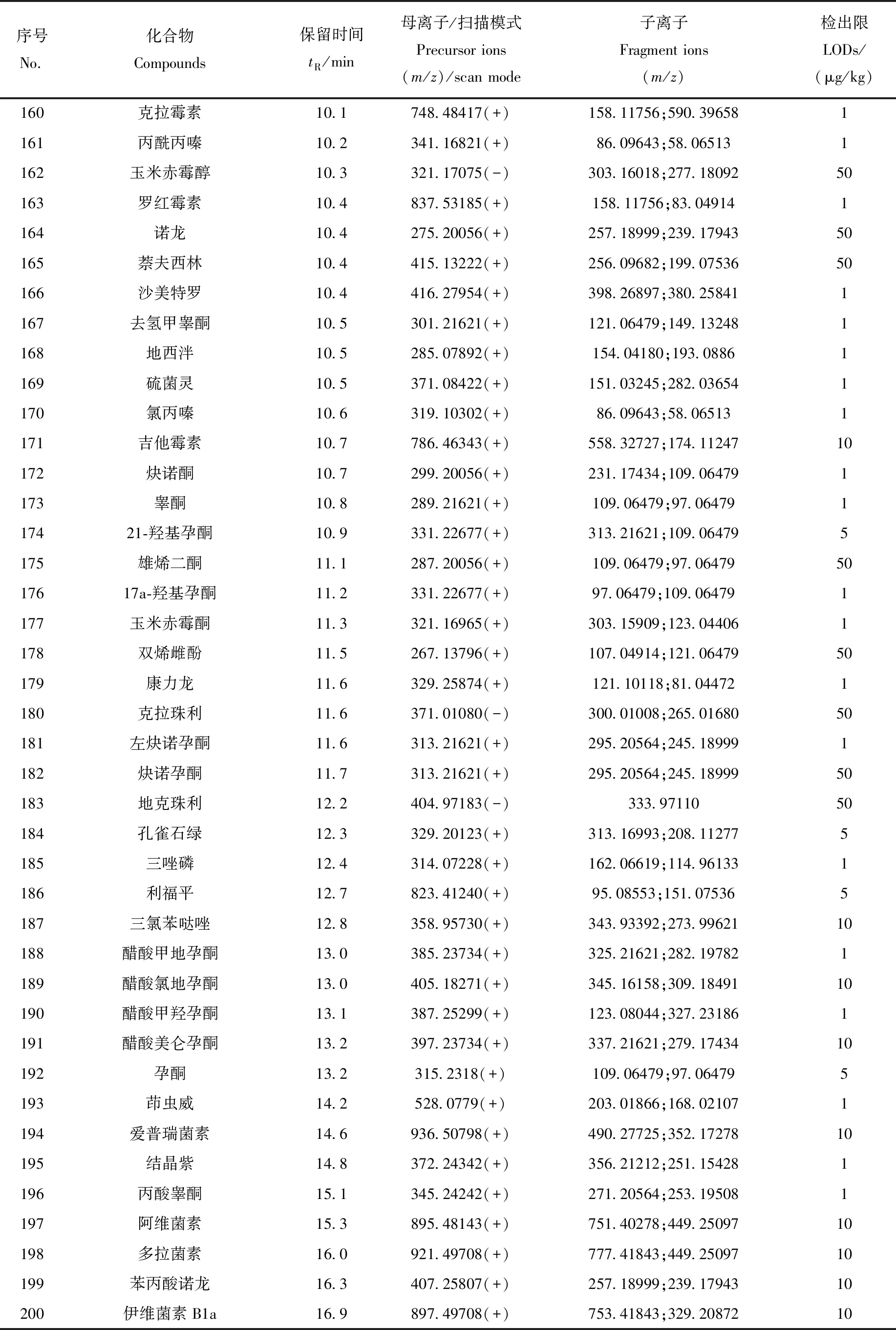
续表1
1.3.6数据库、定性和定量分析 收集200种药物的分子式和相对分子质量信息,在优化的液相色谱和质谱条件下,获取0.1 mg/L各类药物的色谱保留时间、母离子及加合模式,并在梯度碰撞能量(20、50、80 eV)下获得碎片离子信息。利用TraceFinder软件建立含有化合物名称、CAS号、分子式、精确分子质量、保留时间、母离子质荷比(精确至10-5)以及碎片离子质荷比的数据库。
利用Full MS/dd-MS2模式采集数据,结合建立的数据库进行定性筛查,确认检出的标准为:母离子质荷比偏差<3×10-6;保留时间偏差<15 s;同位素质荷比偏差<10×10-6、相对丰度偏差<25%、综合比对>75%;碎片离子质荷比偏差<20×10-6,且至少有1个碎片离子满足此要求。采用基质匹配标准曲线外标法,利用母离子进行定量分析。
2 结果与讨论
2.1 样品前处理的优化
本实验的200种目标物极性差异较大,采用乙腈、乙酸乙酯为提取剂,对草鱼加标样品进行提取实验。结果表明,乙腈作为提取剂时,多数药物能被提取出来,但激素类、镇静剂类和咪唑类药物的提取效率较低;乙酸乙酯作为提取剂时,虽然能够提高激素类和镇静剂类药物的提取效率,但沙星类、β-激动剂类和染料类药物的提取效率较低。为实现不同极性化合物均可达到高效率提取,本实验采用两步提取的方法,即先用乙腈超声提取,再用乙酸乙酯对残渣超声提取,经两次提取,检测回收率为30%~120%,其中,有75%药物的回收率大于50%。
2.2 色谱条件的优化
本实验涉及的目标物种类繁多、性质各异,为达到所有目标物同时分析、准确定性的目的,选择Thermo AccucoreaQ柱作为色谱柱,对200种目标物进行分离实验。结果显示,非极性、弱极性和极性化合物在该色谱柱上的保留性能良好,极性化合物的保留性能优于Thermo Accucore C18 色谱柱。以0.1%甲酸水溶液和0.1%甲酸-乙腈溶液为流动相,优化的梯度洗脱条件见1.3.3节,在最佳色谱分离条件下,各药物均得到有效保留,峰形和灵敏度良好。在22 min内,通过1次进样可实现200种药物的快速筛查分析。200种药物的保留时间已列于表1,部分同分异构体和极性化合物的提取离子流图示于图1。
2.3 质谱条件的优化
根据200种药物的电离性质,选用ESI+和ESI-离子化模式,其中,甲砜霉素、氟苯尼考、氯霉素、玉米赤霉醇、卡拉珠利、地克珠利、头孢呋辛和丫啶黄在负离子模式下响应;其余药物均在正离子模式下响应。采用流动注射方式对200种药物分类进行一级质谱全扫描,质量扫描范围m/z150~1 000,选择信号最强的目标离子作为一级母离子。200种药物母离子、子离子及扫描模式列于表1。
2.4 线性范围和检出限
以鱼、虾空白基质溶液配制基质匹配工作曲线进行定量分析,以消除基质效应对测定结果的影响。基质标准曲线的绘制方法为:以空白基质液配制1~1 000 mg/L系列混合标准溶液,检测得到母离子响应值。以药物的浓度为横坐标,响应值为纵坐标,绘制基质标准曲线。结果表明,每种药物在1 000 mg/L检出限范围内,线性关系良好,相关系数R2≥0.990 0。以能够触发二级质谱扫描的各化合物最小峰面积为评价标准,通过不同加标水平确定方法检出限。本方法的检出限为1~50 μg/kg,其中,有100种药物的检出限为1 μg/kg,有42种药物的检出限为5μg/kg,33种药物的检出限为10 μg/kg,24种药物的检出限为50 μg/kg,各药物的检出限列于表1。
2.5 回收率和精密度
为考察本方法的准确性,对空白鱼、虾样品进行标准添加实验,添加水平分别为1、5、10和50 μg/kg,每个添加水平做5个平行样,按1.3节方法处理和检测,计算不同添加水平下药物的回收率和精密度。结果表明,在不同的加标水平下,200种药物的平均回收率为30%~120%;相对标准偏差在5%~30%之间,其中,RSD为5%~15%、15%~20%和20%~30%的比例分别为70%、10%和20%,且相对标准偏差20%~30%主要分布于低加标水平上。
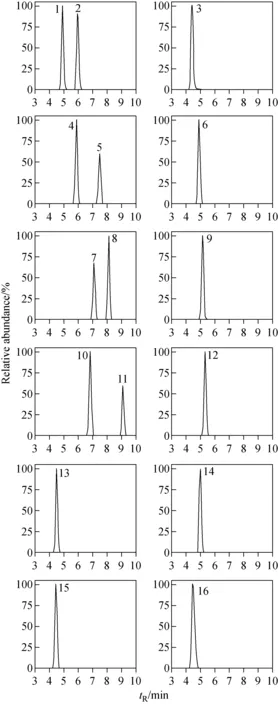
注:1.磺胺二甲异嘧啶;2. 磺胺二甲基嘧啶;3.甲硝唑;4.磺胺二甲噁唑;5.磺胺二甲异噁唑;6.磺胺嘧啶;7.磺胺间二甲氧嘧啶;8. 磺胺邻二甲氧嘧啶;9.多菌灵;10.福莫特罗;11.苯乙醇胺A;12.林可霉素;13.齐帕特罗;14.头孢匹林;15.特布他林;16.喹乙醇图1 混合标准溶液中部分药物的提取离子流图Fig.1 Extracted precursorion chromatograms of some drugs in mix standard solution
3 实际样品筛查
应用本方法对48个鲜活水产品,包括南美白对虾、鳊鱼、草鱼、鲤鱼、鲫鱼和大黄鱼等进行筛查,结果表明,对虾样品中未检出药物残留,鳊鱼、草鱼、鲤鱼、鲫鱼和大黄鱼中均检出药物残留,检出率占样品总量的21%。检出药物的种类包括乙氧喹啉、恩诺沙星、环丙沙星、氧氟沙星、诺氟沙星、甲氧苄啶、磺胺二甲基嘧啶、敌百虫和依维菌素,其中有1个草鱼样品的药物残留较严重,同时检出乙氧喹啉、恩诺沙星、环丙沙星、甲氧苄啶和敌百虫5种药物,含量分别为8.11、363.4、11.7、1 273和26.6 μg/kg,该样品的提取离子流图示于图2。
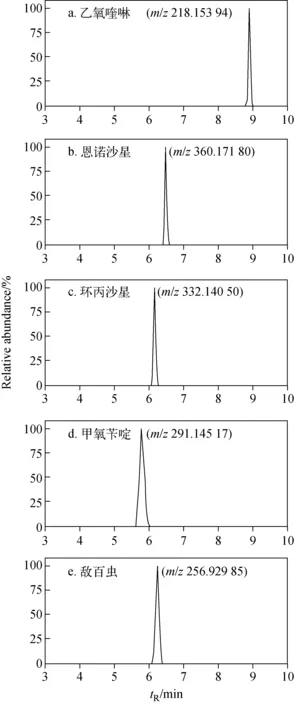
图2 草鱼样品中检出药物的提取离子流图Fig.2 Extracted ion chromatograms of compounds detected in grass carp
4 结论
建立了超高效液相色谱-四极杆/静电场轨道阱高分辨质谱法快速同时筛查鱼、虾样品中200种药物。该方法的样品前处理简单、耗时短、检测效率高、定性准确,可为养殖和流通环节中水产品质量安全监管及隐患排查提供技术支撑。
猜你喜欢
化工设计通讯(2022年10期)2022-12-31
煤化工(2022年3期)2022-07-08
波谱学杂志(2022年2期)2022-06-14
今日农业(2021年4期)2021-11-27
今日农业(2021年15期)2021-11-26
合成纤维工业(2021年5期)2021-10-31
化工设计通讯(2021年5期)2021-05-26
首都食品与医药(2020年1期)2020-10-21
化工设计通讯(2020年11期)2020-01-12
渔业致富指南(2019年21期)2019-11-21
