
IκB-ζ高表达在调控脂多糖诱导细胞炎症反应中的作用
2018-05-14 07:52刘伟张敏龙薄丽艳陈向军李艳燕金发光
中华肺部疾病杂志(电子版) 2018年2期
刘伟 张敏龙 薄丽艳 陈向军 李艳燕 金发光
感染导致的急性肺损伤(acute lung injury, ALI)患病率逐年上升,一旦进展到急性呼吸窘迫综合征(acute respiratory distress syndrome, ARDS)则病死率明显升高[1-4]。如何逆转ALI的“炎症级联反应”是目前的临床难题[5]。IκB-ζ是由NfκBiz基因编码的细胞核内非典型IκB家族蛋白[6],在细胞核内能够双向调节NfκB活性[7]。但是在感染导致的ALI中IκB-ζ起到怎样的炎症调节作用,其调节炎症反应的具体机制如何尚不清楚。本研究通过研究在细胞层面过表达的IκB-ζ蛋白在脂多糖(lipopolysaccharide, LPS)干预的人脐静脉内皮细胞(human umbilical vein endothelial cells, HUVECs)中的作用,旨在明确IκB-ζ在LPS诱导的细胞炎症反应中的具体作用及其具体机制,为感染导致的ALI的救治提供新的思路及理论基础。
材料与方法
一、主要试剂及仪器
HUVECs细胞由第四军医大学病理生理学教研室惠赠,DMEM/F12培养液(Gibco,Cat.No. C11995500BT),10%FBS(四季青),OPTI-MEM(Gibco,Cat.No. 31985062),IkBζ抗体(CST #9244)和IgG抗体(碧云天A7016),NFkB P100/P52抗体(abcam ab175192)。Finnigan Surveyor高效液相系统,AB Sciex TripleTOF®6600质谱仪,Proteinsimple Wes仪器,ABI 7500定量PCR仪。
二、研究方法
1. 细胞培养: HUVECs细胞进行培养,培养条件:DMEM/F12(Gibco,Cat.No. C11995500BT)+10%FBS(四季青),于 37 ℃,5% CO2,90%相对湿度培养箱进行培养,生长至对数期时进行传代。
2. 免疫共沉淀
(1)细胞蛋白提取: 收细胞后,加入400 μl RIPA裂解液,在冰上孵育5 min;漩涡振荡5 s, 12 000×g,4 ℃离心5 min;将上清液(6 mg/ml)转入预冷的离心管中,置于冰上备用,取50 μl蛋白裂解液用于input,1 mg蛋白用于IgG组,1 mg蛋白用于IP组。
(2)免疫共沉淀: 取10 μl抗体原液,分别为IkBζ(cst#9244)抗体和IgG蛋白(碧云天A7016),与1 mg蛋白的裂解液上清混合再用裂解缓冲液稀释至900 μl,在4 ℃缓慢颠倒混匀过夜。取160 μl protein A/G琼脂糖珠(GE Healthcare Life Sciences),加200 μl裂解缓冲液洗涤3次, 4 ℃,3 000 g离心30 s留琼脂糖珠子,加100 μl裂解缓冲液重悬,后进行捕获免疫复合物及洗脱免疫复合物,洗脱后室温下孵育10 min,沸水煮10 min,3 000×g离心30 s,取上清存于-80 ℃,或直接用于Western Blot分析。
(3)SDS-PAGE电泳与凝胶染色: 免疫复合物蛋白测浓度后,在体积分数为10%的SDS-PAGE进行电泳,电泳结束后,用考马斯亮蓝进行凝胶染色。
3. 质谱及数据库分析IκB-ζ蛋白的互作蛋白
(1)蛋白酶解: 将IP及IgG两个蛋白条带分别切胶,进行脱色、还原、烷基化、酶解、脱水、酶切和肽段提取等处理。
(2)质谱检测和数据库分析: 肽段用样品溶解液,通过Finnigan Surveyor高效液相系统让多肽经过C18反向柱进行分离,分离后的肽段直接进入AB Sciex TripleTOF®6600质谱仪进行在线检测。质谱原始文件经过M File Conversion软件处理转换,得到MGF格式文件,然后用MASCOT检索NCBI Rattus数据库和uniprot Rattus数据库进行检索,确定结合蛋白。IP组结合蛋白扣除IgG组结合的非特异蛋白即为特异性结合蛋白。
4. NfκBiz质粒合成: 选用pcDNA3.1作为载体,利用PCR的方法进行质粒合成。PCR引物序列如下: Humo- Nfkbiz -F: 5′ CGGGGTACCATGATTGTGGACAAGCTGC;3′Humo-Nfkbiz-R: 5′ GCCGAATTCCTAATACGGTGGAGCTCTCT 3′。以人CDNA为模板,扩增片段大小为2 175 bp,酶切位点分别为KpnI和EcoRI,前面的碱基为保护碱基。经过提取RNA,PCR,PCR产物纯化,酶切,连接,转化,扩增,挑单克隆等步骤,最终抽提质粒,进行测序后确定质粒制备成功。
5. 细胞转染: 在转染的前1 d,传代准备细胞,按照每孔2×105个细胞接种到12孔板10个,2个作为空白组细胞,4个作为转染空载组,4个作为转染过表达,置于37 ℃、5% CO2培养箱培养,16 h~24 h后待细胞密度生长到60%时即可用于转染。
取50 μl Opti-MEM稀释在含有1.5 ml无血清培养液中,将空载体及Nfkbiz质粒以2 μg/μl浓度分别进行混合,室温静置20 min,然后将混合物分组逐滴均匀加入到对应培养皿中,轻微混匀。置于37 ℃,5% CO2培养箱中培养。4~8 h后换为正常细胞培养基。转染细胞成功后各取2组需要加LPS,分别用LPS(1 μg/ml)干预无血清细胞12 h。48 h后分别做qPCR,Western blot等后续实验。
6. Western blot 分析: 收集细胞后加入80 μl细胞裂解液冰上裂解10 min。以离心半径8 cm,6 000 r/min,离心5 min,收集上清,测定蛋白浓度,制备蛋白样品。Master mix与蛋白样品1︰4的比例充分混匀,漩涡震荡,然后将各个蛋白和Biotinylated Ladder分别插入漂板上进行沸水浴5~10 min。从试剂盒中将装有预制胶的上样板取出,缓慢撕开板上封口薄膜,进行样品、各种试剂的上样,打开Proteinsimple Wes仪器,将样品板放入仪器,从试剂盒中取出毛细管安装在仪器上。双击“compass”程序,单击”Assay”。单击“Start”,程序开始运行,等待实验结果。
7. qPCR检测: 按照RNA提取试剂说明书进行操作,TRNzol Universal。提取RNA后进行逆转录。按照qPCR反应体系进行上机检测,每样品每基因3个重复。设计qPCR引物如下:
IL1-F ATCAGCAAAGAAGTCAAGATGGC
IL1-R CATGGAGTGGGCCATAGCTT
IL6-F CCCACCGGGAACGAAAGAGA
IL6-R GCAGGCAACACCAGGAGCAG
TNF-α-F GCCTGCTGCACTTTGGAGTG
TNF-α-R TCGGGGTTCGAGAAGATGAT
三、统计学方法
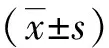
结 果
一、免疫共沉淀所得蛋白质的SDS-PAGE分析
样品经过免疫共沉淀,SDS-PAGE电泳并考马斯亮蓝染色后可见IκB-ζ抗体的IP组与IgG组相比有很多特异性条带。将两组切胶进行质谱分析,检测IκB-ζ的特异性结合蛋白,见图1。

图1 用IκB-ζ抗体对正常HUEVCs细胞全蛋白进行免疫共沉淀的SD-PAGE考马斯亮蓝染色(IP条带为IκB-ζ抗体的免疫沉淀产物,IgG为兔血清做的免疫沉淀产物作为对照)
二、蛋白质谱分析结果显示IκB-ζ与几个重要的蛋白结合
根据数据库分析后挑选出IκB-ζ的结合蛋白TOP23,其中NfκB2(P52,P100)蛋白得分较高,与IκB-ζ互作作用较强,见图2。

图2 IκB-ζ的结合互作蛋白,基因NfκB2编码的蛋白NfκB2(P52,P100)得分较高,与IκB-ζ互作作用较强
三、在LPS干预的HUVECs 细胞中过表达IκB-ζ与NfκB2(P52,P100)亚基结合并抑制其表达
NfκBiz质粒转染HUVECs细胞,转染后检测IκB-ζ蛋白明显高表达,证明NfκBiz质粒合成成功。NfκBiz质粒转染HUVECs细胞后进行LPS干预无血清转染细胞12 h,48 h后检测IκB-ζ,NfκB P100及NfκB P52蛋白含量。结果显示NfκBiz质粒转染的HUVECs细胞进行LPS干预后,IκB-ζ蛋白较LPS组高表达,NfκB P100及NfκB P52较LPS组表达明显降低,见图3、4。

图3 NfκBiz质粒转染HUVECs细胞后IκB-ζ蛋白较对照组明显高表达

图4 NfκBiz质粒转染的HUVECs细胞进行LPS干预后,IκB-ζ蛋白较LPS组高表达,NfκB P100及NfκB P52较LPS组表达明显降低

图5 NfκBiz质粒转染的HUVECs细胞进行LPS干预后,IL-1,TNF-α较LPS组表达下调,IL-6较LPS组表达升高。*与对照组相比差异具有统计学意义(P<0.05),**与LPS组相比差异具有统计学意义(P<0.05)
四、在LPS干预的HUVECs 细胞中IκB-ζ过表达能够下调IL-1,TNF-α表达,同时上调IL-6表达
NfκBiz质粒转染HUVECs细胞后进行LPS干预无血清转染细胞12 h,48 h后检测炎症因子IL-1,IL-6及TNF-α的表达。结果显示NfκBiz质粒转染的HUVECs细胞进行LPS干预后,IL-1,TNF-α较LPS组表达下调,IL-6较LPS组表达升高,见图5。
讨 论
ALI是临床较常见的急危重症,易进展为ARDS,病死率大大升高[8]。感染是导致ALI的常见诱因,ALI的具体发病机制尚未明确[9],但大多数学者认为其发病主要是由于各种原因特别是感染引起的全身炎症反应失控,炎症细胞浸润,炎症因子、自由基大量释放,从而导致非特异性的肺泡-毛细血管上皮的广泛损伤[10]。因此,如何控制细胞及肺组织的炎症级联反应是救治ALI的一大难题。
IκBζ蛋白属于非典型IκB家族蛋白,最早IκBζ是在LPS刺激的巨噬细胞核内发现的,而在细胞浆中未测到该蛋白[11]。有研究表明在巨噬细胞中IκBζ对NF-κB有负反馈抑制作用,抑制IκBζ合成后炎症反应也进一步加重[12-13]。但也有研究表明IκBζ的高表达可以促进炎症反应,炎症因子IL-6表达升高[14- 15],因此,有学者认为IκBζ能够双向调节炎症反应[7]。而本研究发现HUVECs系中LPS干预后IκBζ的高表达能够降低IL-1,TNF-α表达,但同时IL-6表达升高,证明IκBζ确实能够双向调节细胞炎症反应,不仅在巨噬细胞等炎症细胞中,而且在血管内皮细胞中同样能够调节炎症反应。
NF-κB蛋白家族在哺乳动物细胞中存在5个家族成员,包括P50(NF-κB1)、P52(NF-κB2)、RelA(p65)、RelB、c-Rel[16]。每个 NF-κB转录因子家族成员都可形成同源二聚体或与另一个家族成员形成异源二聚体,IκB家族的前体蛋白Pl00和P105与非活化的NF-κB以三聚体的形式存在于细胞中[17]。而IκB家族的前体蛋白Pl00 能够经过泛素化磷酸化加工处理为P52,因此,也有学者将P100与P52合称为NF-κB2蛋白[18]。IκBζ属细胞核IκB蛋白,通过与 NF-κB的亚单位结合,达到抑制细胞核中炎症相关基因的表达[19]。但是在血管内皮细胞的炎症反应中,IκBζ到底是通过结合NF-κB哪个亚单位来调节炎症反应尚不清楚[20-25]。本研究通过蛋白免疫共沉淀找到IκBζ的结合蛋白,再通过蛋白质谱分析找到了多个有意义的结合蛋白,其中发现NF-κB2(P52,P100)蛋白与IκBζ蛋白结合互作,最后又通过过表达IκBζ蛋白后发现P100和P52表达均降低,可能是与过表达的IκBζ蛋白结合的原故[26-30]。
因此,IκBζ可能是通过与Pl00和P52(NF-κB2)的二聚体或者三聚体结合来调节NF-κB的活性,降低IL-1,TNF-α表达,但同时IL-6表达升高,具有双向调节炎症反应的作用。
综上所述,本研究明确了在HUVECs系中IκBζ高表达调节细胞炎症反应的具体作用及机制,为进一步研究IκBζ在LPS导致的ALI中调节炎症反应的机制奠定了基础。
参 考 文 献
1 薄丽艳, 李聪聪, 金发光. 抗氧化治疗在急性肺损伤和急性呼吸窘迫综合征中的应用进展[J/CD]. 中华肺部疾病杂志(电子版), 2016, 9(1): 80-81.
2 吴秀琳, 李明霞, 蔡俊, 等. 转录激活因子3对绿脓杆菌致急性肺损伤小鼠肺泡灌洗液炎症因子的影响[J/CD]. 中华肺部疾病杂志(电子版), 2016, 9(5): 484-488.
3 吴秀琳, 陈复辉, 钱斓兰, 等. 活化转录因子3与急性肺损伤[J/CD]. 中华肺部疾病杂志(电子版), 2017, 10(2): 215-217.
4 Villar J, Sulemanji D, Kacmarek RM. The acute respiratory distress syndrome: incidence and mortality, has it changed?[J]. Curr Opin Crit Care, 2014, 20(1): 3-9.
5 Xu Z, Huang Y, Mao P, et al. Sepsis and ARDS: The Dark Side of Histones[J]. Mediators Inflamm, 2015, 2015: 205054.
6 Yamazaki S, Muta T, Takeshige K. A novel Ikappa B protein, Ikappa B-zeta, induced by proinflammatory stimuli, negatively regulates nuclear factor-kappa B in the nuclei[J]. J Biol Chem, 2001, 276(29): 27657-27662.
7 Motoyama M, Yamazaki S, Eto-Kimura A, et al. Positive and negative regulation of nuclear factor-kappa B-mediated transcription by Ikappa B-zeta, an inducible nuclear protein[J]. J Biol Chem, 2005, 280(9): 7444-7451.
8 Rezoagli E, Fumagalli R, Bellani G. Definition and epidemiology of acute respiratory distress syndrome[J]. Ann Transl Med, 2017, 5(14): 282.
9 Monahan LJ. Acute respiratory distress syndrome[J]. Curr Probl Pediatr Adolesc Health Care, 2013, 43(10): 278-284.
10 Filgueiras LJ, Martins JO, Serezani CH, et al. Sepsis-induced acute lung injury (ALI) is milder in diabetic rats and correlates with impaired NFkB activation[J]. PLoS One, 2012, 7(9): e44987.
11 Kim D, Kolch W, Cho KH. Multiple roles of the NF-kappa B signaling pathway regulated by coupled negative feedback circuits[J]. FASEB J, 2009, 23(9): 2796-2802.
12 Muta T. Ikappa B-zeta: an inducible regulator of nuclear factor-kappa B [J]. Vitam Horm, 2006, 74: 301-316.
13 Muta T, Yamazaki S, Eto A, et al. Ikappa B-zeta, a new anti-inflammatory nuclear protein induced by lipopolysaccharide, is a negative regulator for nuclear factor-kappa B[J]. J Endotoxin Res, 2003, 9(3): 187-191.
14 Seshadri S, Kannan Y, Mitra S, et al. MAIL regulates Human monocyte IL-6 production[J]. J Immunol, 2009, 183(8): 5358-5368.
15 Sundaram K, Rahman MA, Mitra S, et al. IkappaBzeta regulates Human monocyte pro-inflammatory responses induced byStreptococcuspneumoniae[J]. PLoS One, 2016, 11(9): e161931.
16 Rahman A, Fazal F. Blocking NF-kappa B: an inflammatory issue[J]. Proc Am Thorac Soc, 2011, 8(6): 497-503.
17 Mitchell S, Vargas J, Hoffmann A. Signaling via the NFkappaB system[J]. Wiley Interdiscip Rev Syst Biol Med, 2016, 8(3): 227-241.
18 Busino L, Millman SE, Pagano M. SCF-mediated degradation of p100 (NF-kappaB2): mechanisms and relevance in multiple myeloma[J]. Sci Signal, 2012, 5(253): t14.
19 Brennenstuhl H, Armento A, Braczysnki AK, et al. Ikappa Bzeta, an atypical member of the inhibitor of nuclear factor kappa B family, is induced by gamma-irradiation in glioma cells, regulating cytokine secretion and associated with poor prognosis[J]. Int J Oncol, 2015, 47(5): 1971-1980.
20 Zhu J, Weinberg R, Wu X, et al. Ikappab-zeta plays an important role in the ERK-dependent dysregulation of malaria parasite GPI-induced IL-12 expression[J]. IUBMB Life, 2012, 64(2): 187-193.
21 Tartey S, Matsushita K, Vandenbon A, et al. Akirin2 is critical for inducing inflammatory genes by bridging IκB-ζ and the SWI/SNF complex[J]. EMBO J, 2014, 33(20): 2332-2348.
22 Nogai H, Wenzel SS, Hailfinger S, et al. IκB-ζ controls the constitutive NF-κB target gene network and survival of ABC DLBCL[J]. Blood, 2013, 122(13): 2242-2250.
23 Hanihara F, Takahashi Y, Okuma A, et al. Transcriptional and post-transcriptional regulation of IκB-ζ upon engagement of the BCR, TLRs and FcγR[J]. Int Immunol, 2013, 25(9): 531-544.
24 Hörber S, Hildebrand DG, Lieb WS, et al. The Atypical inhibitor of NF-κB, IκBζ, controls macrophage interleukin-10 expression[J]. J Biol Chem, 2016, 291(24): 12851-12861.
25 Hanihara-Tatsuzawa F, Miura H, Kobayashi S, et al. Control of Toll-like receptor-mediated T cell-independent type 1 antibody responses by the inducible nuclear protein IκB-ζ[J]. J Biol Chem, 2014, 289(45): 30925-30936.
26 Hildebrand DG, Alexander E, Hörber S, et al. IκBζ is a transcriptional key regulator of CCL2/MCP-1[J]. J Immunol, 2013, 190(9): 4812-4820.
27 Zhang L, Chen Y, Yue Z, et al. The p65 subunit of NF-κB involves in RIP140-mediated inflammatory and metabolic dysregulation in cardiomyocytes[J]. Arch Biochem Biophys, 2014, 554: 22-27.
28 Pietri JE, Pietri EJ, Potts R, et al. Plasmodium falciparum suppresses the host immune response by inducing the synthesis of insulin-like peptides (ILPs) in the mosquito Anopheles stephensi[J]. Dev Comp Immunol, 2015, 53(1): 134-144.
29 Yunoki T, Tabuchi Y, Hayashi A, et al. BAG3 protects against hyperthermic stress by modulating NF-κB and ERK activities in Human retinoblastoma cells[J]. Graefes Arch Clin Exp Ophthalmol, 2015, 253(3): 399-407.
30 Zhao Q, Du Q, Wei F, et al. An Infectious disease-associated Ⅱ12b polymorphism regulates IL-12/23 p40 transcription involving poly(ADP-ribose) polymerase 1[J]. J Immunol, 2017, 198(7): 2935-2942.
猜你喜欢
——一道江苏高考题的奥秘解读和拓展
中学生物学(2022年7期)2022-09-07
成都医学院学报(2022年4期)2022-08-19
保健医苑(2022年4期)2022-05-05
现代临床医学(2021年4期)2021-07-31
江西农业学报(2021年4期)2021-04-20
汽车维修与保养(2021年8期)2021-02-16
北京大学学报(医学版)(2020年6期)2020-12-14
中国现代医药杂志(2020年10期)2020-12-14
三农资讯半月报(2020年11期)2020-06-21
浙江医学(2017年21期)2017-12-04
