
散发性先天性巨结肠与SOX10基因多态性的关联性
2017-10-12 07:21罗春芬黎胜苗潘志文许嘉川刘征吉於林军
温州医科大学学报 2017年9期
罗春芬,黎胜苗,潘志文,许嘉川,刘征吉,於林军
(1.温州医科大学附属台州医院 小儿外科,浙江 台州 317000;2.浙江大学医学院 细胞生物研究所,浙江 杭州 310058)
散发性先天性巨结肠与SOX10基因多态性的关联性
罗春芬1,黎胜苗1,潘志文2,许嘉川1,刘征吉1,於林军1
(1.温州医科大学附属台州医院 小儿外科,浙江 台州 317000;2.浙江大学医学院 细胞生物研究所,浙江 杭州 310058)
目的:探讨先天性巨结肠(HD)患者SOX10基因多态性与HD发病风险的关系。方法:收集散发性HD患者外周血标本95例为病例组,同时选取健康无便秘史患者80例作为对照组。提取外周血DNA,采用PCR和直接测序法分析SOX10基因多态性在病例组和对照组的分布情况。结果:直接测序发现SOX10基因有4个单核苷酸多态性位点(SNP),这些位点在病例组和对照组中分布差异无统计学意义,关联分析未发现SOX10基因的4个SNP位点与HD的发病风险相关。结论:SOX10基因的4个SNPs位点与散发HD的发病风险没有显著相关性,SOX10基因可能不是散发性HD患者的易感基因。
SOX10基因;先天性巨结肠;多态性,单核苷酸
Abstract: Objective:To determine the association between the variations ofSOX10gene and sporadic HD in Zhejiang province.Methods:Blood from 95 sporadic HD cases and 80 normal cases were collected. Direct sequencing and polymerase chain reaction amplification were used to analyze 4 exons of theSOX10gene for mutations and polymorphisms.Results:In this study, 4 single nucleotide polymorphisms (SNPs) were identified. NoSOX10gene mutations was found with sporadic HD.Conclusion:There is no association between these 4 SNPs of theSOX10gene and HD. TheSOX10gene may not be the susceptibility gene of HD.
Key words:SOX10gene; hirschsprung disease; polymorphism, single nucleotide
先天性巨结肠(hirschsprung disease,HD)又称无神经节细胞性巨结肠(aganglionar megacolon)或肠无神经节细胞症(aganglionosis),是小儿常见的先天性消化道畸形,主要组织病理改变为病变段肠管黏膜下层和肌层神经节细胞缺如,造成该段肠管呈持续性痉挛状态,导致近端肠管代偿性扩张,从而出现便秘、肠梗阻、小肠结肠炎等一系列消化道症状,发病率约为1/5 000[1]。其发病与遗传因素有关,一些与肠神经发育有关的基因突变可导致HD发生。RET基因是主要的易感基因,由RET编码区突变引起的HD占家族性HD的50%和散发性HD的7%~35%;其他基因如EDNRB、EDN3等也与HD发病相关,是HD的易感基因[2-3]。
SOX10基因位于染色体22q13,编码466个氨基酸组成的转录因子,是调控神经嵴细胞发育的关键转录因子[4],主要调节神经嵴源性细胞包括黑色素细胞和肠神经细胞的正常发育[5]。Shah-Waardenburg综合征(WS4)患者中有SOX10基因杂合突变检出,患者除了有HD的症状,还表现为色素缺陷、耳聋等。迄今已发现有多例综合征型HD病例中检测出有携带SOX10基因突变[6-10],但SOX10基因与散发性HD的相关性研究较少。本研究通过对照分析浙江省散发性HD病例和正常人群的SOX10基因多态性,探讨SOX10基因是否与散发性HD的发病相关。
1 材料和方法
1.1 材料
1.1.1 血样收集:收集2011年至2015年间经温州医科大学附属台州医院和浙江大学医学院附属儿童医院临床和病理确诊的95例散发性HD患儿血样为病例组,其中男71例,女24例,年龄2个月~13岁,汉族,按Rome分型标准[11]分为:短段型36例,常见型28例,长段型24例,全结肠型7例。所有病例直系亲属无HD患者,无其他合并症。收集同期健康儿童80例血样为对照组。本研究经医院伦理委员会审核批准,患者监护人知情同意。
1.1.2 PCR引物序列:根据SOX10基因的外显子使用primer5.0软件设计引物,引物序列见表1。

表1 SOX10基因4个外显子引物序列
1.2 方法 取病例组和对照组外周血2 mL,根据DNA提取试剂盒说明进行DNA抽提。PCR反应体系:95 ℃预变性5 min,以94 ℃ 1 min,56~62 ℃(第1外显子56 ℃,第2外显子62 ℃,第3外显子58 ℃,第4外显子56 ℃)30 s,72 ℃ 45 s,循环30次,最后72 ℃延伸10 min。测序之前,聚丙烯酰胺凝胶电泳纯化所有引物,C18柱回收。应用ABI3000自动测序仪进行突变筛查。所有目的基因的测序结果重复3次。
1.3 统计学处理方法 采用SPSS17.0进行统计分析。采用卡方检验检测对照组基因型频率是否符合Hardy-Weinberg平衡,P>0.05表示符合Hardy-Weinberg平衡。运用逻辑回归方法,在显性、共显性、超显性、隐性、加性五种不同遗传模式下分析单个单核苷酸多态性(single nucleotide polymorphisms,SNP)和HD是否相关。P<0.05为差异有统计学意义。
2 结果
通过PCR扩增目的片段后测序,与基因库SOX10序列比对,结果在SOX10基因上发现4个SNPs(见图1)。SNP1在第2外显子编码区,基因序列改变为GAC→GAT;SNP2位于第2外显子编码区,基因序列改变为GGC→GTC,引起第41位甘氨酸变为缬氨酸;SNP3位于第2内含子区域,基因序列改变为C>G;SNP4位于第4外显子编码区,基因序列改变为CAT→CAC,是第309位组氨酸的同义突变。
在对照组中,4个SNP位点的基因型频率均符合Hardy-Weinberg平衡(P值分别为0.39、0.96、0.82、0.39)。基因型分布和4个SNP的等位基因频率在病例组和对照组之间差异均无统计学意义(χ2分别为1.15、0.19、0.38、0.12,P分别为0.28、0.67、0.54、0.73),见表2。
3 讨论
HD发病率约为1/5 000,占先天性消化道畸形疾病的第2位,以肠神经嵴细胞迁移障碍导致肠道局部神经节细胞缺失为主要病理特征,临床上表现为便秘、肠梗阻、小肠结肠炎等消化道症状,严重影响患儿的生活质量及生长发育,严重者甚至危及生命[12]。近年来,HD的发病率呈逐渐上升趋势,其发病机制已成为研究热点。目前认为,HD是由遗传、肠壁内微环境改变、环境等多因素共同作用引起的,而遗传因素最为重要。RET基因及其配体GDNF、NTN,EDNRB基因及其配体EDN3,BMP信号通路,KBP,L1CAM黏附分子,KIAA1279,转录因子PHOX2B、SOX10、MASH1、HAND2和ZFHX1B等在HD发病中起重要作用[13-14],它们的异常引起信号传导的异常,从而进一步影响神经嵴细胞的分化、增殖以及移行,最后导致HD的发生[15-16]。近年来,有关SOX10基因的研究逐渐增多,但未发现相关突变热点,仅对合并有HD相关综合征如WS4(Shah-Waardenburg syndrome,OMIM 277580)及PCWH(peripheral demyelinating neuropathy,central dysmyelinating leukodystrophy,WS,and HD,OMIM 609136)等患者有较多的报道,而对HD患者的研究鲜有报道[6-10]。
SOX10基因位于染色体22q13,属于SOX基因家族成员。SOX基因家族编码一系列SOX家族转录因子,主要特征是含有一个高度保守的HMG-box DNA结合域,参与胚胎发育和细胞分化的调控[17-18]。SOX10基因编码466个氨基酸组成的转录因子,参与调节多能神经嵴细胞的基因表达,在神经嵴细胞增殖、迁移、分化及发育等过程中起着非常重要的作用[5,8,19-20]。PINGAULT等[21]在1998年首先报道了WS4患者存在SOX10基因的突变。2010年,SÁNCHEZMEJÍAS等[10]报道在西班牙HD人群中发现了SOX10缺失(c.153-155del),并且认为在HMG结构域上游的SOX10的缺失在功能上等同于无效突变。本研究选取散发性HD患儿及健康儿童进行病例对照研究,未发现SOX10相关位点的突变与HD发病相关,表明SOX10不是散发性HD患者发病的主要原因。然而,我们在SOX10基因中检测了4个SNP位点,SNP1检测结果与SÁNCHEZ-MEJÍAS等[10]的报道一致,不改变氨基酸编码序列;SNP2是本研究新发现的SNP位点,使得编码的氨基酸从甘氨酸变为缬氨酸,突变位点位于SOX10 HMG保守区域上游,且无一例患者具有WS4、PCWH等其他相关临床表现,此多态性位点在国内尚未见报道,其多态性引起的氨基酸序列对SOX蛋白功能的影响仍不清楚,需要进一步的研究;SNP3是内含子多态性位点;SNP4在SNP数据库中序号是rSl39884,该位点是一个同义突变点,编码氨基酸为组氨酸。在4个位点的分析中,尽管正常对照组符合Hardy-Weinberg平衡,但分析显示4个SNP位点在病例组和对照组中分布无显著性差异,表明这4个SNP位点与HD的发病不存在相关性。由4个SNP位点组成的单体型在HD病例组和正常组中的分布也未见显著性差异,这些单体型与HD发病不具有相关性。以上研究结果表明,SOX10基因可能不是HD发病的易感基因。

图1 SOX10基因测序结果
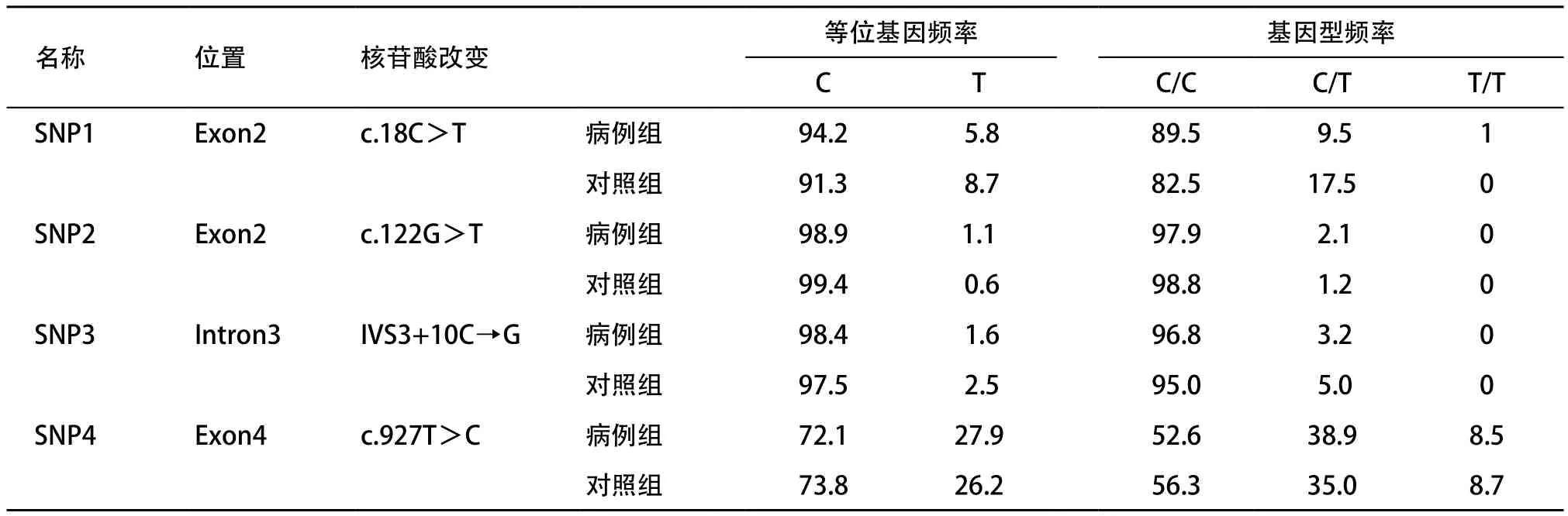
表2 SOX10基因4个SNP位点在病例组和对照组的频率分布(%)
[1] GATH R, GOESSLING A, KELLER K M, et al. Analysis of the RET, GDNF, EDN3, and EDNRB genes in patients with intestinal neuronal dysplasia and Hirschsprung disease[J]. Gut, 2001, 48(5): 671-675.
[2] AURICCHIO A, CASARI G, STAINANO A, et al. Endothelin-B receptor mutations in patients with isolated Hirschsprung disease from a non-inbred population[J]. Hum Mol Genet, 1999, 5(3): 351-354.
[3] BIDAUD C, SALOMON R, VAN CAMP G, et al. Endothelin-3 gene mutations inisolated and syndromic Hirschsprung disease[J]. Eur J Hum Genet, 1997, 5(4): 247-251.
[4] 张艳, 金先庆. 先天性无神经节细胞症的病理学诊断研究进展[J]. 临床小儿外科杂志, 2 0 1 2, 1 1(3): 2 1 6-2 1 8.
[5] BONDURAND N, SHAM M H. The role of SOX10 during enteric nervous system development[J]. Dev Biol, 2013, 382(1): 330-343.
[6] LIANG F, ZHAO M, FAN L, et al. Identification of a de novo mutation of SOX10 in a Chinese patient with Waardenburg syndrome type IV[J]. Int J Pediatr Otorhinolaryngol,2016, 91: 67-71.
[7] JIANG L, CHEN H, JIANG W, et al. Novel mutations in the SOX10 gene in the first two Chinese cases of type IV Waardenburg syndrome[J]. Biochem Biophys Res Commun,2011, 408(4): 620-624.
[8] LECERF L, KAVO A, RUIZ-FERRER M, et al. An impairment of long distance SOX10 regulatory elements underlies isolated Hirschsprung disease[J]. Hum Mutat, 2014, 35(3):303-307.
[9] PINGAULT V, ENTE D, DASTOT-LE MOAL F, et al. Review and update of mutations causing Waardenburg syndrome[J]. Hum Mutat, 2010, 31(4): 391-406.
[10] SÁNCHEZ-MEJÍAS A, WATANABE Y, M FERNÁNDEZ R,et al. Involvement of SOX10 in the pathogenesis of Hirschsprung disease: report of a truncating mutation in an isolated patient[J]. J Mol Med, 2010, 88(5): 507-514.
[11] ROMEO G, RONCHETTO P, LUO Y, et al. Point mutations affecting the tyrosine kinase domain of the RET protooncogene in Hischsprung’s disease[J]. Nature, 1994, 367(6461):377-378.
[12] TJADEN N E, TRAINOR P A. The developmental etiology and pathogenesis of Hirschsprung disease[J]. Transl Res,2013, 162 (1): 1-15.
[13] MCKEOWN S J, STAMP L, HAO M M, et a1. Hirschsprung disease: a developm ental disorder of the enteric nervous system[J]. Wiley Interdiscip Rev Dev Biol, 2013, 2(1): 113-129.
[14] 罗春芬, 於林军, 刘征吉, 等. E D N R B和E D N-3基因多态性与先天性巨结肠的相关性研究[J]. 温州医学院学报, 2 0 1 0,4 0(5): 4 6 2-4 6 8.
[15] BEST K E, GLINIANAIA S V, BYTHELL M, et a1. Hirschsprungs disease in the North of England: prevalence, associated anomalies, and survival[J]. Birth Defects Res A Clin Mol Teratol, 2012, 94(6): 477-480.
[16] 甄亚琴, 徐纪荣. 先天性巨结肠的研究进展[J]. 中国当代医药, 2 0 1 5, 2 2(3 4): 2 1-2 4.
[17] 李颖, 黄英, 王大佳, 等. S o x 1 0和E D N R B在人胚胎肠神经系统中的表达[J]. 中国医科大学学报, 2 0 1 4, 4 3(3): 2 4 3-2 4 6.
[18] BOWLES J, SCHEPERS G, KOOPMAN P. Phylogeny of the SOX family of developmental transcription factors based on sequence and structuralindicators[J]. Dev Biol, 2000, 227(2): 239-255.
[19] BRONNER M E, LEDOUARIN N M. Development and evolution of the neural crest: an overview[J]. Dev Biol,2012, 366(1): 2-9.
[20] SHAM M H, LUI V C, CHEN B L, et al. Novel mutations of SOX10 suggest a dominant negative role in Waardenburg-Shah syndrome[J]. J Med Genet, 2001, 38(9): E30.
[21] PINGAULT V, BONDURAND N, KUHLBRODT K, et al.SOX10 mutations in patients with Waardenburg-Hirschsprung disease[J]. Nat Genet, 1998, 18(2): 171-173.
(本文编辑:丁敏娇)
Association analysis of theSOX10polymorphism and Hirschsprung
LUO Chunfen1, LI Shengmiao1, PANZhiwen2, XU Jiachuan1, LIU Zhengji1, YU Linjun1. 1.Department of Pediatric Surgery, Taizhou Hospital Affiliated to Wenzhou Medical University, Taizhou, 317000; 2.Institute of Cell Biology, Zhejiang University School of Medicine, Hangzhou, 310058
R726.2
A
10.3969/j.issn.2095-9400.2017.09.008
2016-12-26
台州市科技计划项目(121ky08)。
罗春芬(1962-),女,浙江台州人,主任医师。
於林军,副主任医师,Email:yulj@enzemed.com。
猜你喜欢
电子科技大学学报(2022年5期)2022-10-29
天津医科大学学报(2021年4期)2021-08-21
中国生殖健康(2020年4期)2021-01-18
心肺血管病杂志(2020年5期)2021-01-14
中国医药导报(2020年16期)2020-07-31
读天下(2020年2期)2020-04-14
中国生殖健康(2018年4期)2018-11-06
中国当代医药(2015年30期)2015-03-01
湖北农业科学(2014年11期)2014-09-10
现代检验医学杂志(2014年5期)2014-02-02
