
利用退火反应产生粘性末端的基因克隆新方法
2017-08-11 08:51赵婷婷曾静静王启军褚茂平
浙江医学 2017年14期
赵婷婷 曾静静 王启军 褚茂平
利用退火反应产生粘性末端的基因克隆新方法
赵婷婷 曾静静 王启军 褚茂平
目的 通过引物设计和退火反应产生内切酶的粘性末端,旨在开发一种高效、简单、可靠的分子克隆方法。方法 选定限制性内切酶(平末端或者粘性末端)设计3条或4条引物,同时进行两次独立的聚合酶链式反应(PCR),随后把两次PCR产物回收后混在一起经过解链退火,再将其与双酶切的载体质粒进行连接反应,最后转化即可得到目的克隆。 结果 利用此方法成功一次性将若干靶基因(P90rsk、Nprl2、Mios、Ragc、S6k、Pkca)构建在经EcoRV和NotI双酶切后的pcDNA3.1(+)载体上,经酶切及测序验证。 结论 该方法无需考虑靶基因内部是否存在酶切位点,可省去PCR产物酶切及之后的纯化回收过程,可快速并准确地克隆目标基因。
分子克隆 聚合酶链反应 退火 限制性酶切位点 粘性末端
分子克隆是分子生物学实验的常用手段之一。传统的分子克隆利用限制性内切酶切割产生粘性末端以进行目标分子和载体的特异性连接[1]。然而,这种方法具有很大的局限性:当目标片段含有质粒多克隆位点上所有的限制性内切酶时无论选哪一种限制性内切酶都将导致目标DNA片段的切割[2];在进行多个基因克隆时,因各基因序列不同引起的限制性内切酶的选择不一,使得多个基因的克隆变成一个浩大的工程[2]。为了解决传统克隆方法的局限性,此前研究者作出了一定的尝试[3-14],如利用同源重组反应[3-4]和TA克隆方法,两者或增加对重组酶的依赖性或极易出现非特异性突变及二次克隆而不被普遍使用[15-16]。
为了使得分子克隆更加简单和快捷,并减少对限制性内切酶的依赖,本研究设计并验证了一种利用退火产生粘性末端的方法,即根据限制性内切酶酶切所产生的粘性末端情况,设计针对靶基因本身的3条或者4条引物,分两组进行聚合酶链式反应(PCR)反应,将产物进行退火产生粘性末端随后进行传统的分子克隆步骤,现报道如下。
1 材料和方法
1.1 材料 质粒pcDNA3.1(+)由上海市免疫学研究所馈赠。cDNA文库和大肠杆菌DH5α感受态细胞由实验室构建,DNA分子量标准(BM101-01 500μl)购于北京全式金公司,细菌培养基为LB的液体或固体培养基,氨苄青霉素(69-52-3 5g)购于上海生工生物技术有限公司。限制性内切酶EcoRV(R0195S 4000units)、NotI(R0189S 500units)、BamHI(R0136S 10000units)购于美国 NEB公司。T4连接酶(2011A 25 000U)、PCR PrimerSTAR MaxDNA Polymerase(R045Q 50μl×25次)购于日本Takara公司。胶回收试剂盒(ID28704 50次)和质粒抽提试剂盒(ID12362 500次)购于德国QIAGEN公司。PCR仪(BS97MyCyler)购于美国Bio-Rad公司,引物用Oligo Analyzer 1.6软件设计生成。引物合成和基因测序分别由上海生工生物技术有限公司和上海铂尚公司完成。
1.2 方法
1.2.1 引物设计 根据pcDNA3.1(+)上的多克隆位点(MCS),本研究选定EcoRV和NotI,EcoRV 5′-3′序列为GAT∨ATC,酶切后为平末端;NotI5′-3′序列为GC∨GGCCGC,酶切后为粘性末端(5′-3′GGCC),目的基因为P90rsk、Nprl2、Mios、Ragc、S6k和Pkca(表1),用Oligo Analyzer软件设计引物,每个靶基因各设计了3对引物,具体序列及退火温度详见表1。

表1 引物序列表
1.2.2 酶切质粒 选用EcoRV和NotI双酶切pcDNA3.1(+),形成平末端和粘性末端(5′-3′GGCC),1μg质粒需要1μl酶,10×Cutsmart缓冲液2μl,其余用2次蒸馏水(ddH2O)补齐,体系20μl,37℃酶切15min~1h,酶切产物进行胶回收,或可加大酶切体系,-80℃保存酶切后的质粒,可用于后续的分子克隆。
1.2.3 PCR扩增目的片段 提取小鼠胚胎成纤维细胞(MEF)的mRNA反转录构建cDNA文库,以此为模板同时进行2次PCR,加入的1对引物分别是X-F和X-R(X为克隆的目的基因),X-F和GGCCX-R,两者反应体系均为50μl,反应扩增的条件是:98℃变性5min;98℃变性10s;退火温度(表1)30s;72℃延伸(1kb/min依据不同基因长度),经过30个循环,72℃延伸5min后4℃保存。PCR反应结束后,每个样本取5μl用1%琼脂凝胶电泳检测PCR扩增效率,在130V电泳30min,用凝胶成像检测,可以得到2种产物,一种是靶基因全长,另外一种除了保留靶基因全长在其3′末端增加了一段GGCC序列(表2、图1)。PCR结束后,用胶回收试剂盒回收目的条带。
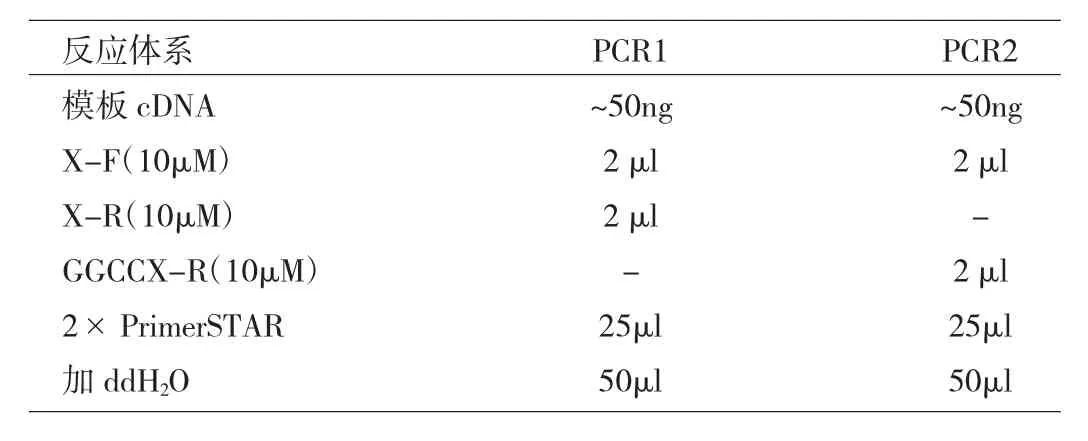
表2 PCR反应体系
1.2.4 退火 可得到2种不同的PCR产物,割胶回收后把2种产物混在一起,经过98℃1min、52℃1min、72℃1min、25℃1min的退火程序,可得到2种不同的产物。
1.2.5 连接 酶切的质粒和退火后的产物摩尔比介于1∶3~1∶10连接效率最高,加入连接酶和连接缓冲液,16~ 25℃连接30min~2h。
1.2.6 转化到大肠杆菌感受态细胞DH5α 连接产物直接转化到感受态细胞DH5α。取全部的连接产物加入到100μl的感受态细胞内,于冰上放置15min,然后42℃热击90s,加入冰育的无抗LB培养基1ml,于摇床37℃转速为220r/min孵育1h,将整个混合液铺到含有抗生素的LB固体培养基,37℃孵育过夜12~16h。
1.2.7 挑取单克隆抽质粒测序鉴定 挑选单克隆2个,接种到含有抗生素的4 ml的LB液体培养基中,转速为220r/min,37℃培养箱孵育过夜12~16h,用质粒提取试剂盒抽提质粒,后续单酶切及测序鉴定。
1.2.8 BamHI单酶切鉴定 取克隆质粒1μg,1μl内切酶BamHI,2×Cutsmart缓冲液5 μl,加ddH2O至10μl体系,37℃酶切15min~1h,酶切产物1%琼脂糖凝胶电泳鉴定,130V30min。
2 结果
2.1 克隆的过程图 见插页图1。
由图1可见,选择真核常用的载体pcDNA3.1(+)以及2个限制性酶切位点EcoRV和NotI,EcoRV 5′-3′序列为GAT∨ATC,产生平末端,NotI 5′-3′序列为GC∨GGCCGC,产生粘性末端,载体转录方向是从EcoRV到NotI,图1酶切后的载体末端是GAT平末端和GGCC粘性末端。根据酶切位点和基因序列设计3条引物:引物1和2基于目的基因序列设计的引物,引物3是基于目的基因3′端加入NotI酶切粘性末端GGCC序列(图1,引物1红箭头,引物2蓝箭头,引物3蓝箭头GGCC)。将引物1和引物2组合,引物1和引物3组合,进行2次独立的以目的基因为模板,分别扩增出目的基因和目的基因3′端增有GGCC序列,1%琼脂糖凝胶电泳跑胶并割胶回收,然后混合两种PCR产物,进行如下反应:98℃1min、52℃1min、72℃1min、25℃1min,模拟解链、退火和延伸过程,形成2种带有粘性末端的产物,只有1种产物能与酶切后的载体匹配,随之将退火后的产物与限制性内切酶切后的质粒常规连接,只有能匹配的片段才能在连接酶作用下形成完整的带有目的片段的重组DNA,随后把连接产物全部转化到感受态细胞DH5α,涂板于含有抗性的固体LB培养基,挑单克隆抽提质粒。
2.2 BamHI单酶切鉴定重组DNA结果 从各个转化平板上挑选2个单克隆,扩增纯化抽提质粒,因pcDNA3.1(+)和基因内部各存在1个BamHI限制性酶切位点,故酶切重组DNA后,若是阳性结果则会有双条带。根据跑胶结果,只有4号泳道的P90rsk质粒未克隆成功(图2),挑选PCR结果正确的质粒送测序。
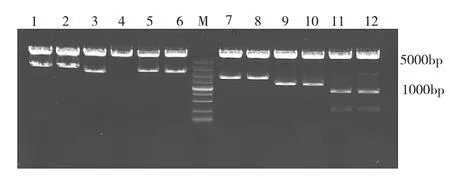
图2 重组质粒的BamHI单酶切结果(M:DNA分子量标准;1~2:Mios;3~4:P90rsk;5~6:Pkca;7~8:S6k;9~10:Ragc:11~12:Nprl2。结果显示只有4号重组DNA未能将基因构建在pcDNA3.1(+),其余均是双条带,表示克隆构建成功)
2.3 基因测序结果 见图3。
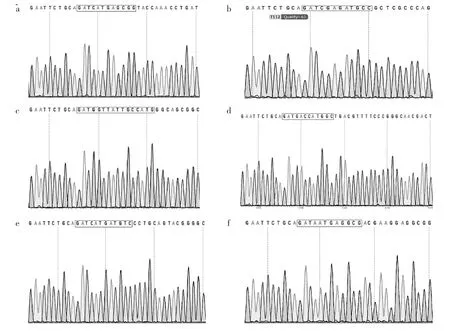
图3 目的基因测序结果(a:Mios;b:P90rsk;c:Nprl2;d:Pkca;e:Ragc;f:S6k)
由图3可见,根据构建的载体上有EcoRV的GAT序列以及设计的引物信息,可以很容易定位到插入的基因片段,Pubmed搜索到全基因序列,依次比对即可。图3方括号括出GAT+5′引物,测序结果显示送测的克隆完全正确。图3挑选其中一个正确测序结果显示,序列中没有碱基突变、碱基缺失、碱基插入。
3 讨论
传统将目的基因进行PCR的产物酶切产生粘性末端的方法常会遇到两大问题:第一,克隆单基因时受到基因内所含限制性内切酶的限制,往往因为基因片段太长会使得包含限制性内切酶的数量和种类也随之增大,当基因含有目标载体多克隆位点上所有的限制性内切酶时,传统的克隆方法即用限制性内切酶切割PCR产物产生粘性末端的方法将不能实现基因克隆的目的;第二,当需要同时克隆多个基因时,因为各个基因序列不一引起的限制性内切酶的选择不一,使得多个基因(即使是几个基因)的克隆变成一个浩大的工程,费时费力,而且不一定都能够成功拿到重组质粒。基于此,通过将粘性末端设计在引物上,通过PCR产物的退火来产生实验所需要的粘性末端,将所有需要克隆的目的基因均一化,不再考虑基因内部所包含的限制性内切酶的多少,只需设计前向和后向引物即可。在多基因同时克隆时,退火产生粘性末端的方法所具有的优势使得多基因单一化,即用同一个模式克隆所有需要的基因:只需要选择质粒多克隆位点上任意两个限制性内切酶,将质粒进行酶切回收,为随后的连接反应作好准备。将对应的限制性内切酶切出的粘性末端序列加到每一个需要克隆的基因引物上,统一进行PCR扩增目标基因与胶回收,然后将成对的PCR产物进行4min的退火反应,即可进行连接,大大降低了克隆的工作量,使得克隆乃至批量克隆成为一件简单的事情。
退火反应也有不足的地方,即1个基因的克隆至少需要3条引物,选择的限制性内切酶所切出的末端都是粘性末端则需要4条引物,相较于传统分子克隆,增加了引物合成的成本。不过随着生物合成技术越来越成熟,合成引物的费用也在逐渐减少,退火产生粘性末端的方法将会是花费少、不费时的克隆选择。
[1] Wilson R H,Morton S K,Deiderick H,et al.Engineered DNA ligases with improved activities in vitro[J].Protein Eng Des Sel, 2013,26:471-478.
[2] Luft J R,Snell E H,Detitta G T.Lessons from high-throughput protein crystallization screening:10 years of practical experience [J].Expert Opin Drug Discov,2011,6:465-480.
[3] Huang J,Yu Z,Li M H,et al.A strategy for seamless cloning of large DNA fragments from Streptomyces[J].Biotechniques,2015, 59:193-194,196,198-200.
[4] MotohashiK.A simple and efficient seamless DNA cloning method using SLiCE from Escherichia coli laboratory strains and its application to SLiP site-directed mutagenesis[J]. BMC Biotechnol,2015,15:47.
[5] Larson C B,Crusemann M,Moore B S.PCR-Independent Method of Transformation-Associated Recombination Reveals the Cosmomycin Biosynthetic Gene Cluster in an Ocean Streptomycete[J].JournalOf NaturalProducts,2017,80:1200-1204.
[6] Matsuo Y,Kishimoto H,Horiuchi T,et al.Simple and effective gap-repair cloning using short tracts of flanking homology in fission yeast[J].BiosciBiotechnolBiochem,2010,74:685-689.
[7] Romano D,Valdetara F,Zambelli P,et al.Cloning the putative gene of vinyl phenol reductase of Dekkera bruxellensis in Saccharomyces cerevisiae[J].Food Microbiol,2017,63:92-100.
[8] Schmid-Burgk J L,Schmidt T,Kaiser V,et al.A ligation-independent cloning technique for high-throughput assembly of transcription activator-like effector genes[J].Nat Biotechnol, 2013,31:76-81.
[9] Thieme F,Marillonnet S.Quick and clean cloning[J].Methods Mol Biol,2014,1116:37-48.
[10] Trehan A,Kielbus M,Czapinski J,et al.REPLACR-mutagenesis,a one-step method for site-directed mutagenesis by recombineering[J].SciRep,2016,6:19121.
[11] Del Prete S,De Luca V,Vullo D,et al.A new procedure for the cloning,expression and purification of the beta-carbonic anhydrase from the pathogenic yeast Malassezia globosa,an anti-dandruff drug target[J].J Enzyme Inhib Med Chem,2016,31: 1156-1161.
[12] Gibson D G,Young L,Chuang R Y,et al.Enzymatic assembly of DNA molecules up to several hundred kilobases[J].Nat Methods,2009,6:343-345.
[13] Krishnamurthy VV,Khamo J S,Cho E,et al.Multiplex gene removal by two-step polymerase chain reactions[J].Anal Biochem,2015,481:7-9.
[14] Scholz J,Besir H,Strasser C,et al.A new method to customize protein expression vectors for fast,efficient and background free parallelcloning[J].BMC Biotechnol,2013,13:12.
[15]Zhou M Y,Gomez-Sanchez C E.Universal TA cloning[J].Curr Issues MolBiol,2000,2:1-7.
[16] Zhou M Y,Clark S E,Gomez-Sanchez C E.Universal cloning method by TAstrategy[J].Biotechniques,1995,19:34-35.
A new method for gene clone producing cohesive terminals by annealingreaction
ZHAO Tingting,ZENG Jingjing,WANG Qijun,et al.Department of Pediatrics,the Second Affiliated Hospital/Yuying Children's Hospital of Wenzhou Medical University,Wenzhou 325027,China
Molecular cloning Polymerase chain reaction Annealing Restriction site Cohesive end
2017-05-05)
(本文编辑:马雯娜)
10.12056/j.issn.1006-2785.2017.39.14.2017-1035
浙江省自然科学基金资助项目(LZ17H250001)
325027 温州医科大学附属第二医院/育婴儿童医院儿科(赵婷婷、曾静静、褚茂平);上海交通大学医学院、上海市免疫学研究所(王启军)
褚茂平,E-mail:chmping@hotmail.com
【 Abstract】 Objective To develop a new effective gene clone method by designing three or four primers to produce cohesive terminals. Methods Two independent PCRs were run based on these designed primers,mixed the two PCR products together,and then went through denaturation and annealing.After that,the mixed products were ligated with the digested vector and the ligation products were transformed into DH5α competent cells. Results The target genes(P90rsk,Nprl2,Mios,Ragc, S6k,Pkca)were constructed on vector pcDNA3.1(+),which were digested with EcoRVand NotI.The constructedgenes were verified by single restriction digestion and sequencing. Conclusion The developed method overcomes the limitation ofrestriction endonuclease sites present in the target genes and can also omit the digestion process of the PCR products and subsequent purification.
猜你喜欢
数学年刊A辑(中文版)(2022年3期)2023-01-05
——一道江苏高考题的奥秘解读和拓展
中学生物学(2022年7期)2022-09-07
成都医学院学报(2022年4期)2022-08-19
苏州科技大学学报(自然科学版)(2021年4期)2021-12-02
中学生数理化·中考版(2021年9期)2021-11-20
江西农业学报(2021年4期)2021-04-20
三农资讯半月报(2020年11期)2020-06-21
皮革制作与环保科技(2020年13期)2020-03-17
会计之友(2018年1期)2018-01-21
中学生天地·高中学习版(2008年1期)2008-02-19
